
Lipid-Centric Approaches in Combating Infectious Diseases: Antibacterials, Antifungals and Antivirals with Lipid-Associated Mechanisms of Action

Abstract
:1. Introduction
2. Antibacterials with Lipid-Associated Mechanisms of Action
2.1. Inhibitors of Membrane Lipid Biosynthesis in Bacteria
2.1.1. Biosynthesis of Fatty Acids of Bacterial Membrane Lipids
2.1.2. Biosynthesis of Head Groups of Bacterial Lipids
2.1.3. Biosynthesis of Lipid A
2.2. Agents with Direct Action on Bacterial Lipid Membranes
3. Antifungal Agents with Lipid-Related Mechanisms of Action
3.1. Inhibition of Biosynthesis of Fungal Cell Membrane Components
3.1.1. Biosynthesis of Fatty Acids of Fungal Membrane Lipids
3.1.2. Biosynthesis of Phospholipid Head Groups
3.1.3. Biosynthesis of Sphingolipids
3.1.4. Ergosterol Synthesis

| Inhibitor | Structure | Enzyme | IC50, μM | References | |
|---|---|---|---|---|---|
| terbinafine |  | ERG1 | C. albicans | 0.03 | [390,392] |
| C. parapsilosis | 0.02–0.04 | [390] | |||
| C. glabrata | 0.137 | [390] | |||
| Trichophyton rubrum | 0.002–0.016 | [390,392] | |||
| A. fumigatus | 0.24 | [390] | |||
| naftifine |  | ERG1 | C. albicans | 1.1 | [390] |
| C. parapsilosis | 0.34 | [390] | |||
| T. rubrum | 0.115 ± 0.030 | [392] | |||
| SDZ 87-469 |  | ERG1 | T. rubrum | 0.020 ± 0.005 | [392] |
| C. albicans | 0.011 | [392] | |||
| tolciclate |  | ERG1 | T. rubrum | 0.028 ± 0.003 | [392] |
| C. albicans | 0.12 | [392] | |||
| tolnaftate |  | ERG1 | T. rubrum | 0.052± 0.009 | [392] |
| C. albicans | 1.04 | [392] | |||
| bifonazole |  | ERG11 | C. albicans | 0.3 | [397] |
| clotrimazole |  | ERG11 | C. albicans | 0.091 | [397] |
| miconazole |  | ERG11 | C. albicans | 0.072 | [397] |
| fluconazole |  | ERG11 | C. albicans | 0.051–0.6 | [397,412] |
| C. neoformans | 0.17 | [413] | |||
| Malassezia globosa | 0.206 ± 0.008 | [414] | |||
| itraconazole |  | ERG11 | C. albicans | 0.039–0.4 | [397,412] |
| C. neoformans | 0.17 | [413] | |||
| M. globosa | 0.188 ± 0.008 | [414] | |||
| voriconazole |  | ERG11 | C. neoformans | 0.17 | [413] |
| VT-1129 |  | ERG11 | C. neoformans | 0.16 | [413] |
| ketoconazole |  | ERG11 | C. albicans | 0.064–0.5 | [397,412] |
| M. globosa | 0.176 ± 0.016 | [414] | |||
| ketaminazole |  | ERG11 | M. globosa | 0.321 ± 0.042 | [414] |
| compound 1a |  | ERG24 | C. albicans | 0.063 | [415] |
| compound 1b |  | ERG24 | C. albicans | 0.016 | [415] |
3.2. Agents with Direct Action on Fungal Lipid Membrane
4. Antivirals Targeting Lipid Envelope
4.1. Disrupting Agents
4.1.1. Photosensitizing Antivirals
| Photosensitizer | Structure | Virus | IC50, µM | Reference |
|---|---|---|---|---|
| hypericin |  | HIV-1 | 0.44 | [466] |
| HSV-1 | 0.006 | [469] | ||
| gymnochrome B |  | dengue | 0.029 | [475] |
| hypocrellin A |  | HSV-1 | 0.015 | [469] |
| hypocrellin B |  | HSV-1 | 0.025 | [469] |
| 5-(perylen-3-yl)ethynyl-2′-deoxy-uridine (dUY11) |  | IVA | 0.097–2.7 | [480,498] |
| HSV-1 | 0.048–0.131 | [479] | ||
| HSV-2 | 0.031–0.055 | [479,480] | ||
| HCV | 0.183–0.187 | [479,480] | ||
| mCMV | 0.037 ± 0.016 | [480] | ||
| SINV | 0.006 ± 0.001 | [480] | ||
| TBEV | 0.024 ± 0.013 | [483,499] | ||
| PIV | 2.2 ± 0.5 | [498] | ||
| RSV | 1.8 ± 0.2 | [498] | ||
| SARS-CoV-2 | 0.2564 | [487] | ||
| 5-(perylen-3-yl)ethynyl-arabino-uridine (aUY11) |  | IVA | 0.078–5.2 | [480,498] |
| HSV-1 | 0.048 ± 0.012 | [479] | ||
| HSV-2 | 0.052 ± 0.003 | [480] | ||
| HCV | 0.107 ± 0.041 | [480] | ||
| mCMV | 0.013 ± 0.004 | [480] | ||
| SINV | 0.011 ± 0.005 | [479] | ||
| TBEV | 0.018 ± 0.010 | [483] | ||
| YFV | 0.0086 ± 0.0007 | [484] | ||
| CHIKV | <0.78 | [484] | ||
| PIV | 1.3 ± 0.3 | [498] | ||
| RSV | 2.3 ± 0.1 | [498] | ||
| SARS-CoV-2 | 0.4058 | [487] | ||
| (5Z)-5-[(5-phenylfuran-2-yl)methylidene]-3-prop-2-enyl-2-sulfanylidene-1,3-thiazolidin-4-one (LJ-001) |  | HIV | 0.133 | [492] |
| Newcastle disease virus | 0.095 | [492] | ||
| Ebola virus | 0.9 | [492] | ||
| IVA | 0.026 | [492] | ||
| Nipah virus | 0.048 | [492] | ||
| Hendra virus | 0.018 | [492] | ||
| Rift valley fever virus | 0.02 | [492] | ||
| Semliki forest virus | 0.537 | [492] | ||
| HSV-1 | 0.02 | [492] | ||
| hCMV | 0.13 | [492] | ||
| VSV | 0.298 | [492] | ||
| (Z) 3-ethyl-5-[5-(2-methoxyphenyl)-furan-2-ylmethylene]oxazolid-ine-2,4-dithione (JL-103) |  | HIV | 0.013 | [492] |
| Newcastle disease virus | 0.004 | [492] | ||
| Ebola virus | 0.185 | [492] | ||
| IVA | 0.002 | [492] | ||
| Nipah virus | 0.004 | [492] | ||
| Hendra viru | 0.0005 | [492] | ||
| Rift valley fever virus | 0.003 | [492] | ||
| Semliki forest virus | 0.044 | [492] | ||
| HSV-1 | 0.002 | [492] | ||
| hCMV | 0.004 | [492] | ||
| VSV | 0.011 | [492] | ||
| 5,15-bis(1,3-dimethylimidazol-2-yl)chlorin (ICH-Me2+) |  | SARS-CoV-2 | 0.12 | [494] |
| pheophorbide a |  | SARS-CoV-2 | 0.18 | [497] |
| MERS-CoV | 0.18 | [497] |
4.1.2. Tweezers
4.1.3. Antimicrobial Peptides
4.2. Fusion Inhibitors Affecting Membrane Fluidity and/or Curvature Stress
5. Conclusions
- (i)
- Due to principal differences in the organization of fatty acid synthase systems in bacteria and mammals, the specific inhibitors of bacterial key enzymes, especially the acetyl-CoA-carboxylase complex, various β-ketoacyl-ACP synthases, different NADPH-dependent reductases, β-hydroxyacyl-ACP dehydrases, and acyl-phosphate:glycerol-3-phosphate acyltransferase, are attractive targets for the development of low-toxicity antibacterials.
- (ii)
- The pathway for the synthesis of the lipid fatty acid tails in fungi is similar to that in mammalian cells and, therefore, is not very promising in the search for potential antifungals.
- (iii)
- The presence of a single fundamental pathway for the synthesis of the phospholipid heads in both prokaryotes and eukaryotes makes the majority of the involved enzymes poor targets for antibiotic therapy in bacterial and fungal infections.
- (iv)
- Many enzymes of the lipopolysaccharide (Kdo2-lipid A) biosynthetic pathway in Gram-negative bacteria (UDP-N-acetylglucosamine acyltransferase, UDP-3-O-(R-3-hydroxyacyl)glucosamine N-acyltransferase, UDP-3-O-(R-3-hydroxyacyl)-N-acetylglucosamine deacetylase, and UDP-diacylglucosamine pyrophosphohydrolase) are identified as targets for antibiotic development.
- (v)
- Sphingolipid biosynthetic pathways are conserved from yeast to humans, and the enzymes cannot serve as targets for low-toxicity antifungals. Some inhibitors of inositol-phosphoceramide synthase demonstrate promisingly low effective concentrations.
- (vi)
- The most effective approach when targeting fungal lipid biosynthesis is to search for inhibitors of enzymes in the ergosterol pathway, especially squalene epoxidase, lanosterol 14α-demethylase, and sterol C14-reductase/sterol C8,7-isomerase.
- (vii)
- A preference given to inhibitors that simultaneously act on two enzymes of the lipid biosynthetic pathway or the combination of inhibitors with agents directly affecting the pathogen membrane should reduce the risk of developing antibiotic resistance in pathogenic strains.
- (viii)
- Natural antimicrobial agents exert their defensive activities via pathogen membrane disruption due to pore formation or the disordering of membrane lipids. Due to the high efficiency of naturally occurring antimicrobial agents, their broad-spectrum antibacterial/antifungal/antiviral effect, and their low rate of resistance in pathogen strains, the use of antimicrobial peptides, lipopeptides, and polyenes is a good anti-infective therapeutic strategy.
- (ix)
- The lipid envelope of viruses should be considered as a target for innovative antivirals, disrupting the membranes of virions or inducing curvature stress and inhibiting viral entry.
Author Contributions
Funding
Conflicts of Interest
References
- Kim, D.; Kim, S.; Kwon, Y.; Kim, Y.; Park, H.; Kwak, K.; Lee, H.; Lee, J.H.; Jang, K.M.; Kim, D.; et al. Structural Insights for β-Lactam Antibiotics. Biomol. Ther. 2023, 31, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Dumas, F.; Haanappel, E. Lipids in infectious diseases—The case of AIDS and tuberculosis. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1636–1647. [Google Scholar] [CrossRef]
- Mochalkin, I.; Miller, J.R.; Narasimhan, L.; Thanabal, V.; Erdman, P.; Cox, P.B.; Prasad, J.V.; Lightle, S.; Huband, M.D.; Stover, C.K. Discovery of antibacterial biotin carboxylase inhibitors by virtual screening and fragment-based approaches. ACS Chem. Biol. 2009, 4, 473–483. [Google Scholar] [CrossRef]
- Cheng, C.C.; Shipps, G.W., Jr.; Yang, Z.; Sun, B.; Kawahata, N.; Soucy, K.A.; Soriano, A.; Orth, P.; Xiao, L.; Mann, P.; et al. Discovery and optimization of antibacterial AccC inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 6507–6514. [Google Scholar] [CrossRef]
- Freiberg, C.; Pohlmann, J.; Nell, P.G.; Endermann, R.; Schuhmacher, J.; Newton, B.; Otteneder, M.; Lampe, T.; Häbich, D.; Ziegelbauer, K. Novel bacterial acetyl coenzyme A carboxylase inhibitors with antibiotic efficacy in vivo. Antimicrob. Agents Chemother. 2006, 50, 2707–2712. [Google Scholar] [CrossRef]
- Khandekar, S.S.; Gentry, D.R.; Van Aller, G.S.; Warren, P.; Xiang, H.; Silverman, C.; Doyle, M.L.; Chambers, P.A.; Konstantinidis, A.K.; Brandt, M.; et al. Identification, substrate specificity, and inhibition of the Streptococcus pneumoniae beta-ketoacyl-acyl carrier protein synthase III (FabH). J. Biol. Chem. 2001, 276, 30024–30030. [Google Scholar] [CrossRef]
- Choi, K.H.; Kremer, L.; Besra, G.S.; Rock, C.O. Identification and substrate specificity of beta -ketoacyl (acyl carrier protein) synthase III (mtFabH) from Mycobacterium tuberculosis. J. Biol. Chem. 2000, 275, 28201–28207. [Google Scholar] [CrossRef] [PubMed]
- Price, A.C.; Choi, K.H.; Heath, R.J.; Li, Z.; White, S.W.; Rock, C.O. Inhibition of beta-ketoacyl-acyl carrier protein synthases by thiolactomycin and cerulenin. Structure and mechanism. J. Biol. Chem. 2001, 276, 6551–6559. [Google Scholar] [CrossRef] [PubMed]
- Tsay, J.T.; Rock, C.O.; Jackowski, S. Overproduction of beta-ketoacyl-acyl carrier protein synthase I imparts thiolactomycin resistance to Escherichia coli K-12. J. Bacteriol. 1992, 174, 508–513. [Google Scholar] [CrossRef]
- Wang, J.; Kodali, S.; Lee, S.H.; Galgoci, A.; Painter, R.; Dorso, K.; Racine, F.; Motyl, M.; Hernandez, L.; Tinney, E.; et al. Discovery of platencin, a dual FabF and FabH inhibitor with in vivo antibiotic properties. Proc. Natl. Acad. Sci. USA 2007, 104, 7612–7616. [Google Scholar] [CrossRef] [PubMed]
- Jayasuriya, H.; Herath, K.B.; Zhang, C.; Zink, D.L.; Basilio, A.; Genilloud, O.; Diez, M.T.; Vicente, F.; Gonzalez, I.; Salazar, O.; et al. Isolation and structure of platencin: A FabH and FabF dual inhibitor with potent broad-spectrum antibiotic activity. Angew. Chem. Int. Ed. Engl. 2007, 46, 4684–4688. [Google Scholar] [CrossRef]
- Wang, J.; Soisson, S.M.; Young, K.; Shoop, W.; Kodali, S.; Galgoci, A.; Painter, R.; Parthasarathy, G.; Tang, Y.S.; Cummings, R.; et al. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature 2006, 441, 358–361. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Rock, C.O. Evaluation of epigallocatechin gallate and related plant polyphenols as inhibitors of the FabG and FabI reductases of bacterial type II fatty-acid synthase. J. Biol. Chem. 2004, 279, 30994–31001. [Google Scholar] [CrossRef]
- Tasdemir, D.; Lack, G.; Brun, R.; Rüedi, P.; Scapozza, L.; Perozzo, R. Inhibition of Plasmodium falciparum fatty acid biosynthesis: Evaluation of FabG, FabZ, and FabI as drug targets for flavonoids. J. Med. Chem. 2006, 49, 3345–3353. [Google Scholar] [CrossRef] [PubMed]
- Belluti, F.; Perozzo, R.; Lauciello, L.; Colizzi, F.; Kostrewa, D.; Bisi, A.; Gobbi, S.; Rampa, A.; Bolognesi, M.L.; Recanatini, M.; et al. Design, synthesis, and biological and crystallographic evaluation of novel inhibitors of Plasmodium falciparum enoyl-ACP-reductase (PfFabI). J. Med. Chem. 2013, 56, 7516–7526. [Google Scholar] [CrossRef] [PubMed]
- Kirmizibekmez, H.; Calis, I.; Perozzo, R.; Brun, R.; Dönmez, A.A.; Linden, A.; Rüedi, P.; Tasdemir, D. Inhibiting activities of the secondary metabolites of Phlomis brunneogaleata against parasitic protozoa and plasmodial enoyl-ACP Reductase, a crucial enzyme in fatty acid biosynthesis. Planta Med. 2004, 70, 711–717. [Google Scholar] [CrossRef]
- Sohn, M.J.; Zheng, C.J.; Kim, W.G. Macrolactin S, a new antibacterial agent with FabG-inhibitory activity from Bacillus sp. AT28. J. Antibiot. 2008, 61, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Bhowruth, V.; Brown, A.K.; Besra, G.S. Synthesis and biological evaluation of NAS-21 and NAS-91 analogues as potential inhibitors of the mycobacterial FAS-II dehydratase enzyme Rv0636. Microbiology 2008, 154, 1866–1875. [Google Scholar] [CrossRef]
- McGillick, B.E.; Kumaran, D.; Vieni, C.; Swaminathan, S. β-Hydroxyacyl-acyl Carrier Protein Dehydratase (FabZ) from Francisella tularensis and Yersinia pestis: Structure Determination, Enzymatic Characterization, and Cross-Inhibition Studies. Biochemistry 2016, 55, 1091–1099. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, L.; Zhang, Y.; Zhang, H.; Du, J.; Ding, J.; Guo, Y.; Jiang, H.; Shen, X. Emodin targets the beta-hydroxyacyl-acyl carrier protein dehydratase from Helicobacter pylori: Enzymatic inhibition assay with crystal structural and thermodynamic characterization. BMC Microbiol. 2009, 9, 91. [Google Scholar] [CrossRef]
- Kumar, V.; Sharma, A.; Pratap, S.; Kumar, P. Biochemical and biophysical characterization of 1,4-naphthoquinone as a dual inhibitor of two key enzymes of type II fatty acid biosynthesis from Moraxella catarrhalis. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.H.; Zhang, L.; Yang, Z.Y.; Han, C.; Hu, L.H.; Jiang, H.L.; Shen, X. Natural product juglone targets three key enzymes from Helicobacter pylori: Inhibition assay with crystal structure characterization. Acta Pharmacol. Sin. 2008, 29, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.J.; Sohn, M.J.; Lee, S.; Kim, W.G. Meleagrin, a new FabI inhibitor from Penicillium chryosogenum with at least one additional mode of action. PLoS ONE 2013, 8, e78922. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Seo, J.H.; Kwak, J.H.; Shin, K.J. Discovery of a potent enoyl-acyl carrier protein reductase (FabI) inhibitor suitable for antistaphylococcal agent. Bioorg. Med. Chem. Lett. 2015, 25, 4481–4486. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.J.; Kim, H.J.; Kim, W.G. Complestatin exerts antibacterial activity by the inhibition of fatty acid synthesis. Biol. Pharm. Bull. 2015, 38, 715–721. [Google Scholar] [CrossRef]
- Surolia, N.; Surolia, A. Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nat. Med. 2001, 7, 167–173. [Google Scholar] [CrossRef]
- Yao, J.; Abdelrahman, Y.M.; Robertson, R.M.; Cox, J.V.; Belland, R.J.; White, S.W.; Rock, C.O. Type II fatty acid synthesis is essential for the replication of Chlamydia trachomatis. J. Biol. Chem. 2014, 289, 22365–22376. [Google Scholar] [CrossRef]
- Yogiara; Mordukhova, E.A.; Kim, D.; Kim, W.G.; Hwang, J.K.; Pan, J.G. The food-grade antimicrobial xanthorrhizol targets the enoyl-ACP reductase (FabI) in Escherichia coli. Bioorg. Med. Chem. Lett. 2020, 30, 127651. [Google Scholar] [CrossRef]
- Cho, J.Y.; Kwon, Y.J.; Sohn, M.J.; Seok, S.J.; Kim, W.G. Phellinstatin, a new inhibitor of enoyl-ACP reductase produced by the medicinal fungus Phellinus linteus. Bioorg. Med. Chem. Lett. 2011, 21, 1716–1718. [Google Scholar] [CrossRef]
- Kim, Y.J.; Sohn, M.J.; Kim, W.G. Chalcomoracin and moracin C, new inhibitors of Staphylococcus aureus enoyl-acyl carrier protein reductase from Morus alba. Biol. Pharm. Bull. 2012, 35, 791–795. [Google Scholar] [CrossRef]
- Kwon, Y.J.; Fang, Y.; Xu, G.H.; Kim, W.G. Aquastatin A, a new inhibitor of enoyl-acyl carrier protein reductase from Sporothrix sp. FN611. Biol. Pharm. Bull. 2009, 32, 2061–2064. [Google Scholar] [CrossRef]
- Zheng, C.J.; Sohn, M.J.; Kim, W.G. Atromentin and leucomelone, the first inhibitors specific to enoyl-ACP reductase (FabK) of Streptococcus pneumoniae. J. Antibiot. 2006, 59, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Grimes, K.D.; Lu, Y.J.; Zhang, Y.M.; Luna, V.A.; Hurdle, J.G.; Carson, E.I.; Qi, J.; Kudrimoti, S.; Rock, C.O.; Lee, R.E. Novel acyl phosphate mimics that target PlsY, an essential acyltransferase in gram-positive bacteria. ChemMedChem 2008, 3, 1936–1945. [Google Scholar] [CrossRef] [PubMed]
- Cherian, P.T.; Yao, J.; Leonardi, R.; Maddox, M.M.; Luna, V.A.; Rock, C.O.; Lee, R.E. Acyl-sulfamates target the essential glycerol-phosphate acyltransferase (PlsY) in Gram-positive bacteria. Bioorg. Med. Chem. 2012, 20, 4985–4994. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Duan, Y.; Zhou, B.; Guo, Q.; Wang, H.; Hang, X.; Zeng, L.; Jia, J.; Bi, H. The Cyclopropane Fatty Acid Synthase Mediates Antibiotic Resistance and Gastric Colonization of Helicobacter pylori. J. Bacteriol. 2019, 201, e00374-19. [Google Scholar] [CrossRef]
- Parsons, J.B.; Rock, C.O. Bacterial lipids: Metabolism and membrane homeostasis. Prog. Lipid Res. 2013, 52, 249–276. [Google Scholar] [CrossRef]
- Larson, E.C.; Lim, A.L.; Pond, C.D.; Craft, M.; Čavužić, M.; Waldrop, G.L.; Schmidt, E.W.; Barrows, L.R. Pyrrolocin C and equisetin inhibit bacterial acetyl-CoA carboxylase. PLoS ONE 2020, 15, e0233485. [Google Scholar] [CrossRef] [PubMed]
- Freiberg, C.; Brunner, N.A.; Schiffer, G.; Lampe, T.; Pohlmann, J.; Brands, M.; Raabe, M.; Häbich, D.; Ziegelbauer, K. Identification and characterization of the first class of potent bacterial acetyl-CoA carboxylase inhibitors with antibacterial activity. J. Biol. Chem. 2004, 279, 26066–26073. [Google Scholar] [CrossRef] [PubMed]
- Freiberg, C.; Fischer, H.P.; Brunner, N.A. Discovering the mechanism of action of novel antibacterial agents through transcriptional profiling of conditional mutants. Antimicrob. Agents Chemother. 2005, 49, 749–759. [Google Scholar] [CrossRef]
- Pohlmann, J.; Lampe, T.; Shimada, M.; Nell, P.G.; Pernerstorfer, J.; Svenstrup, N.; Brunner, N.A.; Schiffer, G.; Freiberg, C. Pyrrolidinedione derivatives as antibacterial agents with a novel mode of action. Bioorg. Med. Chem. Lett. 2005, 15, 1189–1192. [Google Scholar] [CrossRef]
- Toor, H.G.; Banerjee, D.I.; Chauhan, J.B. In Silico Evaluation of Human Cathelicidin LL-37 as a Novel Therapeutic Inhibitor of Panton-Valentine Leukocidin Toxin of Methicillin-Resistant Staphylococcus aureus. Microb. Drug Resist. 2021, 27, 602–615. [Google Scholar] [CrossRef]
- Liu, X.; Fortin, P.D.; Walsh, C.T. Andrimid producers encode an acetyl-CoA carboxyltransferase subunit resistant to the action of the antibiotic. Proc. Natl. Acad. Sci. USA 2008, 105, 13321–13326. [Google Scholar] [CrossRef]
- Silvers, M.A.; Robertson, G.T.; Taylor, C.M.; Waldrop, G.L. Design, synthesis, and antibacterial properties of dual-ligand inhibitors of acetyl-CoA carboxylase. J. Med. Chem. 2014, 57, 8947–8959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wei, S.; Wu, W. Preliminary studies on the antibacterial mechanism of Yanglingmycin. Pestic. Biochem. Physiol. 2018, 147, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Kuldeep, J.; Sharma, S.K.; Singh, B.N.; Siddiqi, M.I. Computational exploration and anti-mycobacterial activity of potential inhibitors of Mycobacterium tuberculosis acetyl coenzyme A carboxylase as anti-tubercular agents. SAR QSAR Environ. Res. 2021, 32, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Sharma, A.; Pratap, S.; Kumar, P. Biophysical and in silico interaction studies of aporphine alkaloids with Malonyl-CoA: ACP transacylase (FabD) from drug resistant Moraxella catarrhalis. Biochimie 2018, 149, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Heath, R.J.; Rock, C.O. Inhibition of beta-ketoacyl-acyl carrier protein synthase III (FabH) by acyl-acyl carrier protein in Escherichia coli. J. Biol. Chem. 1996, 271, 10996–11000. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H.; Heath, R.J.; Rock, C.O. Beta-ketoacyl-acyl carrier protein synthase III (FabH) is a determining factor in branched-chain fatty acid biosynthesis. J. Bacteriol. 2000, 182, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Choudhry, A.E.; Janson, C.A.; Grooms, M.; Daines, R.A.; Lonsdale, J.T.; Khandekar, S.S. Crystal structure and substrate specificity of the beta-ketoacyl-acyl carrier protein synthase III (FabH) from Staphylococcus aureus. Protein Sci. 2005, 14, 2087–2094. [Google Scholar] [CrossRef]
- Musayev, F.; Sachdeva, S.; Scarsdale, J.N.; Reynolds, K.A.; Wright, H.T. Crystal structure of a substrate complex of Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein synthase III (FabH) with lauroyl-coenzyme A. J. Mol. Biol. 2005, 346, 1313–1321. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, Y.S.; Fu, J.; Zhu, H.L. Novel FabH inhibitors: A patent and article literature review (2000—2012). Expert. Opin. Ther. Pat. 2012, 22, 1325–1336. [Google Scholar] [CrossRef]
- Wallace, K.K.; Lobo, S.; Han, L.; McArthur, H.A.; Reynolds, K.A. In vivo and In vitro effects of thiolactomycin on fatty acid biosynthesis in Streptomyces collinus. J. Bacteriol. 1997, 179, 3884–3891. [Google Scholar] [CrossRef]
- Han, L.; Lobo, S.; Reynolds, K.A. Characterization of beta-ketoacyl-acyl carrier protein synthase III from Streptomyces glaucescens and its role in initiation of fatty acid biosynthesis. J. Bacteriol. 1998, 180, 4481–4486. [Google Scholar] [CrossRef]
- Alhamadsheh, M.M.; Waters, N.C.; Huddler, D.P.; Kreishman-Deitrick, M.; Florova, G.; Reynolds, K.A. Synthesis and biological evaluation of thiazolidine-2-one 1,1-dioxide as inhibitors of Escherichia coli beta-ketoacyl-ACP-synthase III (FabH). Bioorg. Med. Chem. Lett. 2007, 17, 879–883. [Google Scholar] [CrossRef]
- Li, H.Q.; Shi, L.; Li, Q.S.; Liu, P.G.; Luo, Y.; Zhao, J.; Zhu, H.L. Synthesis of C(7) modified chrysin derivatives designing to inhibit beta-ketoacyl-acyl carrier protein synthase III (FabH) as antibiotics. Bioorg. Med. Chem. 2009, 17, 6264–6269. [Google Scholar] [CrossRef] [PubMed]
- Lv, P.C.; Wang, K.R.; Yang, Y.; Mao, W.J.; Chen, J.; Xiong, J.; Zhu, H.L. Design, synthesis and biological evaluation of novel thiazole derivatives as potent FabH inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 6750–6754. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Q.; Luo, Y.; Lv, P.C.; Shi, L.; Liu, C.H.; Zhu, H.L. Design and synthesis of novel deoxybenzoin derivatives as FabH inhibitors and anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2010, 20, 2025–2028. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Zheng, Q.Z.; Qian, Y.; Shi, L.; Zhao, J.; Zhu, H.L. Synthesis, antibacterial activities and molecular docking studies of peptide and Schiff bases as targeted antibiotics. Bioorg. Med. Chem. 2009, 17, 7861–7871. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Fang, R.Q.; Zhu, Z.W.; Yang, Y.; Cheng, K.; Zhong, W.Q.; Zhu, H.L. Design and synthesis of potent inhibitors of beta-ketoacyl-acyl carrier protein synthase III (FabH) as potential antibacterial agents. Eur. J. Med. Chem. 2010, 45, 4358–4364. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Zheng, Q.Z.; Hou, J.; Zhou, Y.; Liu, C.H.; Zhao, J.; Zhu, H.L. Synthesis, molecular modeling and biological evaluation of PSB as targeted antibiotics. Bioorg. Med. Chem. 2010, 18, 2447–2455. [Google Scholar] [CrossRef] [PubMed]
- Lv, P.C.; Sun, J.; Luo, Y.; Yang, Y.; Zhu, H.L. Design, synthesis, and structure-activity relationships of pyrazole derivatives as potential FabH inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 4657–4660. [Google Scholar] [CrossRef]
- Zhang, H.J.; Zhu, D.D.; Li, Z.L.; Sun, J.; Zhu, H.L. Synthesis, molecular modeling and biological evaluation of β-ketoacyl-acyl carrier protein synthase III (FabH) as novel antibacterial agents. Bioorg. Med. Chem. 2011, 19, 4513–4519. [Google Scholar] [CrossRef]
- Li, H.Q.; Luo, Y.; Zhu, H.L. Discovery of vinylogous carbamates as a novel class of β-ketoacyl-acyl carrier protein synthase III (FabH) inhibitors. Bioorg. Med. Chem. 2011, 19, 4454–4459. [Google Scholar] [CrossRef]
- Li, Z.L.; Li, Q.S.; Zhang, H.J.; Hu, Y.; Zhu, D.D.; Zhu, H.L. Design, synthesis and biological evaluation of urea derivatives from o-hydroxybenzylamines and phenylisocyanate as potential FabH inhibitors. Bioorg. Med. Chem. 2011, 19, 4413–4420. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, L.R.; Hu, Y.; Zhang, S.; Fu, J.; Wang, X.M.; Zhu, H.L. Synthesis and antimicrobial activities of oximes derived from O-benzylhydroxylamine as FabH inhibitors. ChemMedChem 2012, 7, 1587–1593. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Du, Q.R.; Sun, J.; Li, J.R.; Fang, F.; Li, D.D.; Qian, Y.; Gong, H.B.; Zhao, J.; Zhu, H.L. Novel Schiff-base-derived FabH inhibitors with dioxygenated rings as antibiotic agents. ChemMedChem 2013, 8, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Zhang, Y.B.; Tang, J.F.; Yang, Y.S.; Chen, R.Q.; Zhang, F.; Zhu, H.L. Design, synthesis and antibacterial activities of vanillic acylhydrazone derivatives as potential β-ketoacyl-acyl carrier protein synthase III (FabH) inhibitors. Eur. J. Med. Chem. 2012, 57, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luo, Y.; Hu, Y.; Zhu, D.D.; Zhang, S.; Liu, Z.J.; Gong, H.B.; Zhu, H.L. Design, synthesis and antimicrobial activities of nitroimidazole derivatives containing 1,3,4-oxadiazole scaffold as FabH inhibitors. Bioorg. Med. Chem. 2012, 20, 4316–4322. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.S.; Zhang, F.; Gao, C.; Zhang, Y.B.; Wang, X.L.; Tang, J.F.; Sun, J.; Gong, H.B.; Zhu, H.L. Discovery and modification of sulfur-containing heterocyclic pyrazoline derivatives as potential novel class of β-ketoacyl-acyl carrier protein synthase III (FabH) inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 4619–4624. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, C.P.; Ma, H.P.; Zhao, M.Y.; Xue, Y.R.; Wang, X.M.; Zhu, H.L. Design, synthesis and antimicrobial activities evaluation of Schiff base derived from secnidazole derivatives as potential FabH inhibitors. Bioorg. Med. Chem. 2013, 21, 3120–3126. [Google Scholar] [CrossRef] [PubMed]
- Li, J.R.; Li, D.D.; Wang, R.R.; Sun, J.; Dong, J.J.; Du, Q.R.; Fang, F.; Zhang, W.M.; Zhu, H.L. Design and synthesis of thiazole derivatives as potent FabH inhibitors with antibacterial activity. Eur. J. Med. Chem. 2014, 75, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.T.; Wang, Z.C.; Sang, Y.L.; Tao, X.X.; Teraiya, S.B.; Wang, P.F.; Wen, Q.; Zhou, X.J.; Ding, L.; Yang, Y.H.; et al. Design and synthesis of 2-styryl of 5-Nitroimidazole derivatives and antimicrobial activities as FabH inhibitors. Eur. J. Med. Chem. 2014, 76, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Yang, Y.; Zhao, J.; Chen, Y. Synthesis and antibacterial activity of cinnamaldehyde acylhydrazone with a 1,4-benzodioxan fragment as a novel class of potent β-ketoacyl-acyl carrier protein synthase III (FabH) inhibitor. Chem. Pharm. Bull. 2014, 62, 1110–1118. [Google Scholar] [CrossRef]
- Segretti, N.D.; Serafim, R.A.; Segretti, M.C.; Miyata, M.; Coelho, F.R.; Augusto, O.; Ferreira, E.I. New antibacterial agents: Hybrid bioisoster derivatives as potential E. coli FabH inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3988–3993. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, Y.S.; Song, X.D.; Lu, L.; Zhu, H.L. Study of Schiff-Base-Derived with Dioxygenated Rings and Nitrogen Heterocycle as Potential β-Ketoacyl-acyl Carrier Protein Synthase III (FabH) Inhibitors. Chem. Pharm. Bull. 2017, 65, 178–185. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Reynolds, K.A. Purification, characterization, and identification of novel inhibitors of the beta-ketoacyl-acyl carrier protein synthase III (FabH) from Staphylococcus aureus. Antimicrob. Agents Chemother. 2002, 46, 1310–1318. [Google Scholar] [CrossRef]
- He, X.; Reeve, A.M.; Desai, U.R.; Kellogg, G.E.; Reynolds, K.A. 1,2-dithiole-3-ones as potent inhibitors of the bacterial 3-ketoacyl acyl carrier protein synthase III (FabH). Antimicrob. Agents Chemother. 2004, 48, 3093–3102. [Google Scholar] [CrossRef]
- Pishchany, G.; Mevers, E.; Ndousse-Fetter, S.; Horvath, D.J., Jr.; Paludo, C.R.; Silva-Junior, E.A.; Koren, S.; Skaar, E.P.; Clardy, J.; Kolter, R. Amycomicin is a potent and specific antibiotic discovered with a targeted interaction screen. Proc. Natl. Acad. Sci. USA 2018, 115, 10124–10129. [Google Scholar] [CrossRef]
- Singh, S.; Soni, L.K.; Gupta, M.K.; Prabhakar, Y.S.; Kaskhedikar, S.G. QSAR studies on benzoylaminobenzoic acid derivatives as inhibitors of beta-ketoacyl-acyl carrier protein synthase III. Eur. J. Med. Chem. 2008, 43, 1071–1080. [Google Scholar] [CrossRef]
- Nie, Z.; Perretta, C.; Lu, J.; Su, Y.; Margosiak, S.; Gajiwala, K.S.; Cortez, J.; Nikulin, V.; Yager, K.M.; Appelt, K.; et al. Structure-based design, synthesis, and study of potent inhibitors of beta-ketoacyl-acyl carrier protein synthase III as potential antimicrobial agents. J. Med. Chem. 2005, 48, 1596–1609. [Google Scholar] [CrossRef]
- Ashek, A.; Cho, S.J. A combined approach of docking and 3D QSAR study of beta-ketoacyl-acyl carrier protein synthase III (FabH) inhibitors. Bioorg. Med. Chem. 2006, 14, 1474–1482. [Google Scholar] [CrossRef]
- Jones, P.B.; Parrish, N.M.; Houston, T.A.; Stapon, A.; Bansal, N.P.; Dick, J.D.; Townsend, C.A. A new class of antituberculosis agents. J. Med. Chem. 2000, 43, 3304–3314. [Google Scholar] [CrossRef] [PubMed]
- Alhamadsheh, M.M.; Musayev, F.; Komissarov, A.A.; Sachdeva, S.; Wright, H.T.; Scarsdale, N.; Florova, G.; Reynolds, K.A. Alkyl-CoA disulfides as inhibitors and mechanistic probes for FabH enzymes. Chem. Biol. 2007, 14, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhong, W.; Li, R.J.; Li, S. Synthesis of potent inhibitors of β-ketoacyl-acyl carrier protein synthase III as potential antimicrobial agents. Molecules 2012, 17, 4770–4781. [Google Scholar] [CrossRef]
- Borgaro, J.G.; Chang, A.; Machutta, C.A.; Zhang, X.; Tonge, P.J. Substrate recognition by β-ketoacyl-ACP synthases. Biochemistry 2011, 50, 10678–10686. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Jia, J.; Edwards, P.; Dehesh, K.; Schneider, G.; Lindqvist, Y. Crystal structure of beta-ketoacyl-acyl carrier protein synthase II from E.coli reveals the molecular architecture of condensing enzymes. EMBO J. 1998, 17, 1183–1191. [Google Scholar] [CrossRef]
- Qiu, X.; Janson, C.A.; Konstantinidis, A.K.; Nwagwu, S.; Silverman, C.; Smith, W.W.; Khandekar, S.; Lonsdale, J.; Abdel-Meguid, S.S. Crystal structure of beta-ketoacyl-acyl carrier protein synthase III. A key condensing enzyme in bacterial fatty acid biosynthesis. J. Biol. Chem. 1999, 274, 36465–36471. [Google Scholar] [CrossRef]
- Olsen, J.G.; Kadziola, A.; von Wettstein-Knowles, P.; Siggaard-Andersen, M.; Lindquist, Y.; Larsen, S. The X-ray crystal structure of beta-ketoacyl [acyl carrier protein] synthase I. FEBS Lett. 1999, 460, 46–52. [Google Scholar] [CrossRef]
- Davies, C.; Heath, R.J.; White, S.W.; Rock, C.O. The 1.8 A crystal structure and active-site architecture of beta-ketoacyl-acyl carrier protein synthase III (FabH) from Escherichia coli. Structure 2000, 8, 185–195. [Google Scholar] [CrossRef]
- Heath, R.J.; White, S.W.; Rock, C.O. Inhibitors of fatty acid synthesis as antimicrobial chemotherapeutics. Appl. Microbiol. Biotechnol. 2002, 58, 695–703. [Google Scholar] [CrossRef]
- Bommineni, G.R.; Kapilashrami, K.; Cummings, J.E.; Lu, Y.; Knudson, S.E.; Gu, C.; Walker, S.G.; Slayden, R.A.; Tonge, P.J. Thiolactomycin-Based Inhibitors of Bacterial β-Ketoacyl-ACP Synthases with in Vivo Activity. J. Med. Chem. 2016, 59, 5377–5390. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, J.D.; Dong, L.B.; Manoogian, K.; Shen, B. Biosynthetic Origin of the Ether Ring in Platensimycin. J. Am. Chem. Soc. 2016, 138, 16711–16721. [Google Scholar] [CrossRef] [PubMed]
- Manallack, D.T.; Crosby, I.T.; Khakham, Y.; Capuano, B. Platensimycin: A promising antimicrobial targeting fatty acid synthesis. Curr. Med. Chem. 2008, 15, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, G.A.I.; Nojima, S.; Yamano, Y.; Aono, A.; Arai, M.; Mitarai, S.; Tanaka, T.; Yoshimitsu, T. Potent growth inhibitory activity of (±)-platencin towards multi-drug-resistant and extensively drug-resistant Mycobacterium tuberculosis. Med. Chem. Commun. 2013, 4, 720–723. [Google Scholar] [CrossRef]
- Brown, A.K.; Taylor, R.C.; Bhatt, A.; Fütterer, K.; Besra, G.S. Platensimycin activity against mycobacterial beta-ketoacyl-ACP synthases. PLoS ONE 2009, 4, e6306. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Sakha Ghosh, P.; Manna, K. A Review on Platensimycin: A Selective FabF Inhibitor. Int. J. Med. Chem. 2016, 2016, 9706753. [Google Scholar] [CrossRef] [PubMed]
- Martens, E.; Demain, A.L. Platensimycin and platencin: Promising antibiotics for future application in human medicine. J. Antibiot. 2011, 64, 705–710. [Google Scholar] [CrossRef]
- Shang, R.; Liang, J.; Yi, Y.; Liu, Y.; Wang, J. Review of Platensimycin and Platencin: Inhibitors of β-Ketoacyl-acyl Carrier Protein (ACP) Synthase III (FabH). Molecules 2015, 20, 16127–16141. [Google Scholar] [CrossRef]
- Su, M.; Qiu, L.; Deng, Y.; Ruiz, C.H.; Rudolf, J.D.; Dong, L.B.; Feng, X.; Cameron, M.D.; Shen, B.; Duan, Y.; et al. Evaluation of Platensimycin and Platensimycin-Inspired Thioether Analogues against Methicillin-Resistant Staphylococcus aureus in Topical and Systemic Infection Mouse Models. Mol. Pharm. 2019, 16, 3065–3071. [Google Scholar] [CrossRef]
- Deng, Y.; Weng, X.; Li, Y.; Su, M.; Wen, Z.; Ji, X.; Ren, N.; Shen, B.; Duan, Y.; Huang, Y. Late-Stage Functionalization of Platensimycin Leading to Multiple Analogues with Improved Antibacterial Activity In vitro and in Vivo. J. Med. Chem. 2019, 62, 6682–6693. [Google Scholar] [CrossRef]
- Feng, Z.; Chakraborty, D.; Dewell, S.B.; Reddy, B.V.; Brady, S.F. Environmental DNA-encoded antibiotics fasamycins A and B inhibit FabF in type II fatty acid biosynthesis. J. Am. Chem. Soc. 2012, 134, 2981–2987. [Google Scholar] [CrossRef]
- Zheng, Z.; Parsons, J.B.; Tangallapally, R.; Zhang, W.; Rock, C.O.; Lee, R.E. Discovery of novel bacterial elongation condensing enzyme inhibitors by virtual screening. Bioorg. Med. Chem. Lett. 2014, 24, 2585–2588. [Google Scholar] [CrossRef]
- Wickramasinghe, S.R.; Inglis, K.A.; Urch, J.E.; Müller, S.; van Aalten, D.M.; Fairlamb, A.H. Kinetic, inhibition and structural studies on 3-oxoacyl-ACP reductase from Plasmodium falciparum, a key enzyme in fatty acid biosynthesis. Biochem. J. 2006, 393, 447–457. [Google Scholar] [CrossRef]
- Kristan, K.; Bratkovic, T.; Sova, M.; Gobec, S.; Prezelj, A.; Urleb, U. Novel inhibitors of beta-ketoacyl-ACP reductase from Escherichia coli. Chem. Biol. Interact. 2009, 178, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Vella, P.; Rudraraju, R.S.; Lundbäck, T.; Axelsson, H.; Almqvist, H.; Vallin, M.; Schneider, G.; Schnell, R. A FabG inhibitor targeting an allosteric binding site inhibits several orthologs from Gram-negative ESKAPE pathogens. Bioorg. Med. Chem. 2021, 30, 115898. [Google Scholar] [CrossRef] [PubMed]
- Cukier, C.D.; Hope, A.G.; Elamin, A.A.; Moynie, L.; Schnell, R.; Schach, S.; Kneuper, H.; Singh, M.; Naismith, J.H.; Lindqvist, Y.; et al. Discovery of an allosteric inhibitor binding site in 3-Oxo-acyl-ACP reductase from Pseudomonas aeruginosa. ACS Chem. Biol. 2013, 8, 2518–2527. [Google Scholar] [CrossRef]
- Hu, J.; Webster, D.; Cao, J.; Shao, A. The safety of green tea and green tea extract consumption in adults—Results of a systematic review. Regul. Toxicol. Pharmacol. 2018, 95, 412–433. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, A.A.; Efimova, S.S.; Ostroumova, O.S. Lipid Microenvironment Modulates the Pore-Forming Ability of Polymyxin B. Antibiotics 2022, 11, 1445. [Google Scholar] [CrossRef]
- Chernyshova, D.N.; Tyulin, A.A.; Ostroumova, O.S.; Efimova, S.S. Discovery of the Potentiator of the Pore-Forming Ability of Lantibiotic Nisin: Perspectives for Anticancer Therapy. Membranes 2022, 12, 1166. [Google Scholar] [CrossRef] [PubMed]
- Efimova, S.S.; Malykhina, A.I.; Ostroumova, O.S. Triggering the Amphotericin B Pore-Forming Activity by Phytochemicals. Membranes 2023, 13, 670. [Google Scholar] [CrossRef]
- Sharma, S.K.; Kapoor, M.; Ramya, T.N.; Kumar, S.; Kumar, G.; Modak, R.; Sharma, S.; Surolia, N.; Surolia, A. Identification, characterization, and inhibition of Plasmodium falciparum beta-hydroxyacyl-acyl carrier protein dehydratase (FabZ). J. Biol. Chem. 2003, 278, 45661–45671. [Google Scholar] [CrossRef]
- He, L.; Zhang, L.; Liu, X.; Li, X.; Zheng, M.; Li, H.; Yu, K.; Chen, K.; Shen, X.; Jiang, H.; et al. Discovering potent inhibitors against the beta-hydroxyacyl-acyl carrier protein dehydratase (FabZ) of Helicobacter pylori: Structure-based design, synthesis, bioassay, and crystal structure determination. J. Med. Chem. 2009, 52, 2465–2481. [Google Scholar] [CrossRef]
- Saling, S.C.; Comar, J.F.; Mito, M.S.; Peralta, R.M.; Bracht, A. Actions of juglone on energy metabolism in the rat liver. Toxicol. Appl. Pharmacol. 2011, 257, 319–327. [Google Scholar] [CrossRef]
- Zhu, K.; Choi, K.H.; Schweizer, H.P.; Rock, C.O.; Zhang, Y.M. Two aerobic pathways for the formation of unsaturated fatty acids in Pseudomonas aeruginosa. Mol. Microbiol. 2006, 60, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Leesong, M.; Henderson, B.S.; Gillig, J.R.; Schwab, J.M.; Smith, J.L. Structure of a dehydratase-isomerase from the bacterial pathway for biosynthesis of unsaturated fatty acids: Two catalytic activities in one active site. Structure 1996, 4, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.P.; DeMendoza, D.; Polacco, M.L.; Cronan, J.E., Jr. Beta-hydroxydecanoyl thio ester dehydrase does not catalyze a rate-limiting step in Escherichia coli unsaturated fatty acid synthesis. Biochemistry 1983, 22, 5897–5902. [Google Scholar] [CrossRef] [PubMed]
- Marrakchi, H.; Choi, K.H.; Rock, C.O. A new mechanism for anaerobic unsaturated fatty acid formation in Streptococcus pneumoniae. J. Biol. Chem. 2002, 277, 44809–44816. [Google Scholar] [CrossRef]
- Fozo, E.M.; Quivey, R.G., Jr. The fabM gene product of Streptococcus mutans is responsible for the synthesis of monounsaturated fatty acids and is necessary for survival at low pH. J. Bacteriol. 2004, 186, 4152–4158. [Google Scholar] [CrossRef]
- Wang, H.; Cronan, J.E. Functional replacement of the FabA and FabB proteins of Escherichia coli fatty acid synthesis by Enterococcus faecalis FabZ and FabF homologues. J. Biol. Chem. 2004, 279, 34489–34495. [Google Scholar] [CrossRef]
- Bi, H.; Wang, H.; Cronan, J.E. FabQ, a dual-function dehydratase/isomerase, circumvents the last step of the classical fatty acid synthesis cycle. Chem. Biol. 2013, 20, 1157–1167. [Google Scholar] [CrossRef]
- Altabe, S.G.; Aguilar, P.; Caballero, G.M.; de Mendoza, D. The Bacillus subtilis acyl lipid desaturase is a delta5 desaturase. J. Bacteriol. 2003, 185, 3228–3231. [Google Scholar] [CrossRef]
- Moynié, L.; Leckie, S.M.; McMahon, S.A.; Duthie, F.G.; Koehnke, A.; Taylor, J.W.; Alphey, M.S.; Brenk, R.; Smith, A.D.; Naismith, J.H. Structural insights into the mechanism and inhibition of the β-hydroxydecanoyl-acyl carrier protein dehydratase from Pseudomonas aeruginosa. J. Mol. Biol. 2013, 425, 365–377. [Google Scholar] [CrossRef]
- Moynié, L.; Hope, A.G.; Finzel, K.; Schmidberger, J.; Leckie, S.M.; Schneider, G.; Burkart, M.D.; Smith, A.D.; Gray, D.W.; Naismith, J.H. A Substrate Mimic Allows High-Throughput Assay of the FabA Protein and Consequently the Identification of a Novel Inhibitor of Pseudomonas aeruginosa FabA. J. Mol. Biol. 2016, 428, 108–120. [Google Scholar] [CrossRef] [PubMed]
- McMurry, L.M.; Oethinger, M.; Levy, S.B. Triclosan targets lipid synthesis. Nature 1998, 394, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Perozzo, R.; Kuo, M.; Sidhu, A.; Valiyaveettil, J.T.; Bittman, R.; Jacobs, W.R., Jr.; Fidock, D.A.; Sacchettini, J.C. Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl acyl carrier protein reductase. J. Biol. Chem. 2002, 277, 13106–13114. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, S.; Sullivan, T.J.; Johnson, F.; Novichenok, P.; Cui, G.; Simmerling, C.; Tonge, P.J. Inhibition of the bacterial enoyl reductase FabI by triclosan: A structure-reactivity analysis of FabI inhibition by triclosan analogues. J. Med. Chem. 2004, 47, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Yoon, Y.M.; Jung, S.J.; Yun, I.N.; Kim, C.M.; Kim, J.M.; Kwak, J.H. CG400462, a new bacterial enoyl-acyl carrier protein reductase (FabI) inhibitor. Int. J. Antimicrob. Agents 2007, 30, 446–451. [Google Scholar] [CrossRef]
- Park, H.S.; Yoon, Y.M.; Jung, S.J.; Kim, C.M.; Kim, J.M.; Kwak, J.H. Antistaphylococcal activities of CG400549, a new bacterial enoyl-acyl carrier protein reductase (FabI) inhibitor. J. Antimicrob. Chemother. 2007, 60, 568–574. [Google Scholar] [CrossRef]
- Sampson, P.B.; Picard, C.; Handerson, S.; McGrath, T.E.; Domagala, M.; Leeson, A.; Romanov, V.; Awrey, D.E.; Thambipillai, D.; Bardouniotis, E.; et al. Spiro-naphthyridinone piperidines as inhibitors of S. aureus and E. coli enoyl-ACP reductase (FabI). Bioorg. Med. Chem. Lett. 2009, 19, 5355–5358. [Google Scholar] [CrossRef]
- Ramnauth, J.; Surman, M.D.; Sampson, P.B.; Forrest, B.; Wilson, J.; Freeman, E.; Manning, D.D.; Martin, F.; Toro, A.; Domagala, M.; et al. 2,3,4,5-Tetrahydro-1H-pyrido[2,3-b and e][1,4]diazepines as inhibitors of the bacterial enoyl ACP reductase, FabI. Bioorg. Med. Chem. Lett. 2009, 19, 5359–5362. [Google Scholar] [CrossRef]
- Escaich, S.; Prouvensier, L.; Saccomani, M.; Durant, L.; Oxoby, M.; Gerusz, V.; Moreau, F.; Vongsouthi, V.; Maher, K.; Morrissey, I.; et al. The MUT056399 inhibitor of FabI is a new antistaphylococcal compound. Antimicrob. Agents Chemother. 2011, 55, 4692–4697. [Google Scholar] [CrossRef]
- Banevicius, M.A.; Kaplan, N.; Hafkin, B.; Nicolau, D.P. Pharmacokinetics, pharmacodynamics and efficacy of novel FabI inhibitor AFN-1252 against MSSA and MRSA in the murine thigh infection model. J. Chemother. 2013, 25, 26–31. [Google Scholar] [CrossRef]
- Schiebel, J.; Chang, A.; Shah, S.; Lu, Y.; Liu, L.; Pan, P.; Hirschbeck, M.W.; Tareilus, M.; Eltschkner, S.; Yu, W.; et al. Rational design of broad spectrum antibacterial activity based on a clinically relevant enoyl-acyl carrier protein (ACP) reductase inhibitor. J. Biol. Chem. 2014, 289, 15987–16005. [Google Scholar] [CrossRef]
- Mandal, S.; Parish, T. A Novel Benzoxaborole Is Active against Escherichia coli and Binds to FabI. Antimicrob. Agents Chemother. 2021, 65, e0262220. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Maxwell, J.B.; Rock, C.O. Resistance to AFN-1252 arises from missense mutations in Staphylococcus aureus enoyl-acyl carrier protein reductase (FabI). J. Biol. Chem. 2013, 288, 36261–36271. [Google Scholar] [CrossRef]
- Mehboob, S.; Song, J.; Hevener, K.E.; Su, P.C.; Boci, T.; Brubaker, L.; Truong, L.; Mistry, T.; Deng, J.; Cook, J.L.; et al. Structural and biological evaluation of a novel series of benzimidazole inhibitors of Francisella tularensis enoyl-ACP reductase (FabI). Bioorg. Med. Chem. Lett. 2015, 25, 1292–1296. [Google Scholar] [CrossRef] [PubMed]
- Takahata, S.; Iida, M.; Yoshida, T.; Kumura, K.; Kitagawa, H.; Hoshiko, S. Discovery of 4-Pyridone derivatives as specific inhibitors of enoyl-acyl carrier protein reductase (FabI) with antibacterial activity against Staphylococcus aureus. J. Antibiot. 2007, 60, 123–128. [Google Scholar] [CrossRef]
- Wang, S.F.; Yin, Y.; Wu, X.; Qiao, F.; Sha, S.; Lv, P.C.; Zhao, J.; Zhu, H.L. Synthesis, molecular docking and biological evaluation of coumarin derivatives containing piperazine skeleton as potential antibacterial agents. Bioorg. Med. Chem. 2014, 22, 5727–5737. [Google Scholar] [CrossRef]
- Hu, Y.; Shen, Y.; Wu, X.; Tu, X.; Wang, G.X. Synthesis and biological evaluation of coumarin derivatives containing imidazole skeleton as potential antibacterial agents. Eur. J. Med. Chem. 2018, 143, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Mistry, T.; Ren, J.; Johnson, M.E.; Mehboob, S. A novel series of enoyl reductase inhibitors targeting the ESKAPE pathogens, Staphylococcus aureus and Acinetobacter baumannii. Bioorg. Med. Chem. 2018, 26, 65–76. [Google Scholar] [CrossRef]
- Davis, M.C.; Franzblau, S.G.; Martin, A.R. Syntheses and evaluation of benzodiazaborine compounds against M. tuberculosis H37Rv In vitro. Bioorg. Med. Chem. Lett. 1998, 8, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.; Saffon, N.; Sammartino, J.C.; Degiacomi, G.; Pasca, M.R.; Lherbet, C. First triclosan-based macrocyclic inhibitors of InhA enzyme. Bioorg. Chem. 2020, 95, 103498. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, U.H.; Rao, S.P.S.; Kondreddi, R.R.; Noble, C.G.; Camacho, L.R.; Tan, B.H.; Ng, S.H.; Ng, P.S.; Ma, N.L.; Lakshminarayana, S.B.; et al. Direct inhibitors of InhA are active against Mycobacterium tuberculosis. Sci. Transl. Med. 2015, 7, 269ra3. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Madhavapeddi, P.; Naik, M.; Murugan, K.; Shinde, V.; Nandishaiah, R.; Bhat, J.; Kumar, A.; Hameed, S.; Holdgate, G.; et al. Methyl-thiazoles: A novel mode of inhibition with the potential to develop novel inhibitors targeting InhA in Mycobacterium tuberculosis. J. Med. Chem. 2013, 56, 8533–8542. [Google Scholar] [CrossRef]
- Šink, R.; Sosič, I.; Živec, M.; Fernandez-Menendez, R.; Turk, S.; Pajk, S.; Alvarez-Gomez, D.; Lopez-Roman, E.M.; Gonzales-Cortez, C.; Rullas-Triconado, J.; et al. Design, synthesis, and evaluation of new thiadiazole-based direct inhibitors of enoyl acyl carrier protein reductase (InhA) for the treatment of tuberculosis. J. Med. Chem. 2015, 58, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.D.; Dixit, S.R.; Kulkarni, V.H.; Lherbet, C.; Nadagouda, M.N.; Aminabhavi, T.M. Synthesis, biological evaluation and in silico molecular modeling of pyrrolyl benzohydrazide derivatives as enoyl ACP reductase inhibitors. Eur. J. Med. Chem. 2017, 126, 286–297. [Google Scholar] [CrossRef]
- Rotta, M.; Pissinate, K.; Villela, A.D.; Back, D.F.; Timmers, L.F.; Bachega, J.F.; de Souza, O.N.; Santos, D.S.; Basso, L.A.; Machado, P. Piperazine derivatives: Synthesis, inhibition of the Mycobacterium tuberculosis enoyl-acyl carrier protein reductase and SAR studies. Eur. J. Med. Chem. 2015, 90, 436–447. [Google Scholar] [CrossRef]
- Chollet, A.; Mori, G.; Menendez, C.; Rodriguez, F.; Fabing, I.; Pasca, M.R.; Madacki, J.; Korduláková, J.; Constant, P.; Quémard, A.; et al. Design, synthesis and evaluation of new GEQ derivatives as inhibitors of InhA enzyme and Mycobacterium tuberculosis growth. Eur. J. Med. Chem. 2015, 101, 218–235. [Google Scholar] [CrossRef]
- Hartkoorn, R.C.; Sala, C.; Neres, J.; Pojer, F.; Magnet, S.; Mukherjee, R.; Uplekar, S.; Boy-Röttger, S.; Altmann, K.H.; Cole, S.T. Towards a new tuberculosis drug: Pyridomycin—Nature’s isoniazid. EMBO Mol. Med. 2012, 4, 1032–1042. [Google Scholar] [CrossRef]
- Karioti, A.; Skaltsa, H.; Zhang, X.; Tonge, P.J.; Perozzo, R.; Kaiser, M.; Franzblau, S.G.; Tasdemir, D. Inhibiting enoyl-ACP reductase (FabI) across pathogenic microorganisms by linear sesquiterpene lactones from Anthemis auriculata. Phytomedicine 2008, 15, 1125–1129. [Google Scholar] [CrossRef]
- Brenwald, N.P.; Fraise, A.P. Triclosan resistance in methicillin-resistant Staphylococcus aureus (MRSA). J. Hosp. Infect. 2003, 55, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.J.; Kim, J.A.; Pan, J.G. Signature gene expression profile of triclosan-resistant Escherichia coli. J. Antimicrob. Chemother. 2010, 65, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Ciusa, M.L.; Furi, L.; Knight, D.; Decorosi, F.; Fondi, M.; Raggi, C.; Coelho, J.R.; Aragones, L.; Moce, L.; Visa, P.; et al. A novel resistance mechanism to triclosan that suggests horizontal gene transfer and demonstrates a potential selective pressure for reduced biocide susceptibility in clinical strains of Staphylococcus aureus. Int. J. Antimicrob. Agents 2012, 40, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Chuanchuen, R.; Beinlich, K.; Hoang, T.T.; Becher, A.; Karkhoff-Schweizer, R.R.; Schweizer, H.P. Cross-resistance between triclosan and antibiotics in Pseudomonas aeruginosa is mediated by multidrug efflux pumps: Exposure of a susceptible mutant strain to triclosan selects nfxB mutants overexpressing MexCD-OprJ. Antimicrob. Agents Chemother. 2001, 45, 428–432. [Google Scholar] [CrossRef]
- Wang, L.; Mao, B.; He, H.; Shang, Y.; Zhong, Y.; Yu, Z.; Yang, Y.; Li, H.; An, J. Comparison of hepatotoxicity and mechanisms induced by triclosan (TCS) and methyl-triclosan (MTCS) in human liver hepatocellular HepG2 cells. Toxicol. Res. 2018, 8, 38–45. [Google Scholar] [CrossRef]
- Marrakchi, H.; Dewolf, W.E., Jr.; Quinn, C.; West, J.; Polizzi, B.J.; So, C.Y.; Holmes, D.J.; Reed, S.L.; Heath, R.J.; Payne, D.J.; et al. Characterization of Streptococcus pneumoniae enoyl-(acyl-carrier protein) reductase (FabK). Biochem. J. 2003, 370, 1055–1062. [Google Scholar] [CrossRef]
- Heath, R.J.; Rock, C.O. A triclosan-resistant bacterial enzyme. Nature 2000, 406, 145–146. [Google Scholar] [CrossRef]
- Heath, R.J.; Su, N.; Murphy, C.K.; Rock, C.O. The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J. Biol. Chem. 2000, 275, 40128–40133. [Google Scholar] [CrossRef]
- Huang, Y.H.; Lin, J.S.; Ma, J.C.; Wang, H.H. Functional Characterization of Triclosan-Resistant Enoyl-acyl-carrier Protein Reductase (FabV) in Pseudomonas aeruginosa. Front. Microbiol. 2016, 7, 1903. [Google Scholar] [CrossRef]
- Kim, S.H.; Khan, R.; Choi, K.; Lee, S.W.; Rhee, S. A triclosan-resistance protein from the soil metagenome is a novel enoyl-acyl carrier protein reductase: Structure-guided functional analysis. FEBS J. 2020, 287, 4710–4728. [Google Scholar] [CrossRef]
- Seefeld, M.A.; Miller, W.H.; Newlander, K.A.; Burgess, W.J.; DeWolf, W.E., Jr.; Elkins, P.A.; Head, M.S.; Jakas, D.R.; Janson, C.A.; Keller, P.M.; et al. Indole naphthyridinones as inhibitors of bacterial enoyl-ACP reductases FabI and FabK. J. Med. Chem. 2003, 46, 1627–1635. [Google Scholar] [CrossRef]
- Takahata, S.; Iida, M.; Osaki, Y.; Saito, J.; Kitagawa, H.; Ozawa, T.; Yoshida, T.; Hoshiko, S. AG205, a novel agent directed against FabK of Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2006, 50, 2869–2871. [Google Scholar] [CrossRef] [PubMed]
- Mahfuz, A.M.U.B.; Stambuk Opazo, F.; Aguilar, L.F.; Iqbal, M.N. Carfilzomib as a potential inhibitor of NADH-dependent enoyl-acyl carrier protein reductases of Klebsiella pneumoniae and Mycobacterium tuberculosis as a drug target enzyme: Insights from molecular docking and molecular dynamics. J. Biomol. Struct. Dyn. 2022, 40, 4021–4037. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Rock, C.O. Thematic review series: Glycerolipids. Acyltransferases in bacterial glycerophospholipid synthesis. J. Lipid Res. 2008, 49, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- De Mendoza, D.; Klages Ulrich, A.; Cronan, J.E., Jr. Thermal regulation of membrane fluidity in Escherichia coli. Effects of overproduction of beta-ketoacyl-acyl carrier protein synthase I. J. Biol. Chem. 1983, 258, 2098–2101. [Google Scholar] [CrossRef]
- Cronan, J.E., Jr.; Weisberg, L.J.; Allen, R.G. Regulation of membrane lipid synthesis in Escherichia coli. Accumulation of free fatty acids of abnormal length during inhibition of phospholipid synthesis. J. Biol. Chem. 1975, 250, 5835–5840. [Google Scholar] [CrossRef]
- Bell, R.M. Mutants of Escherichia coli defective in membrane phospholipid synthesis. Properties of wild type and Km defective sn-glycerol-3-phosphate acyltransferase activities. J. Biol. Chem. 1975, 250, 7147–7152. [Google Scholar] [CrossRef]
- Lu, Y.J.; Zhang, Y.M.; Grimes, K.D.; Qi, J.; Lee, R.E.; Rock, C.O. Acyl-phosphates initiate membrane phospholipid synthesis in Gram-positive pathogens. Mol. Cell 2006, 23, 765–772. [Google Scholar] [CrossRef]
- Greenway, D.L.; Silbert, D.F. Altered acyltransferase activity in Escherichia coli associated with mutations in acyl coenzyme A synthetase. J. Biol. Chem. 1983, 258, 13034–13042. [Google Scholar] [CrossRef]
- Brinster, S.; Lamberet, G.; Staels, B.; Trieu-Cuot, P.; Gruss, A.; Poyart, C. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature 2009, 458, 83–86. [Google Scholar] [CrossRef]
- Morvan, C.; Halpern, D.; Kénanian, G.; Hays, C.; Anba-Mondoloni, J.; Brinster, S.; Kennedy, S.; Trieu-Cuot, P.; Poyart, C.; Lamberet, G.; et al. Environmental fatty acids enable emergence of infectious Staphylococcus aureus resistant to FASII-targeted antimicrobials. Nat. Commun. 2016, 7, 12944. [Google Scholar] [CrossRef] [PubMed]
- Gloux, K.; Guillemet, M.; Soler, C.; Morvan, C.; Halpern, D.; Pourcel, C.; Vu Thien, H.; Lamberet, G.; Gruss, A. Clinical Relevance of Type II Fatty Acid Synthesis Bypass in Staphylococcus aureus. Antimicrob. Agents Chemother. 2017, 61, e02515-16. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Lee, J.K. Effect of changes in the composition of cellular fatty acids on membrane fluidity of Rhodobacter sphaeroides. J. Microbiol. Biotechnol. 2015, 25, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Grogan, D.W.; Cronan, J.E., Jr. Characterization of Escherichia coli mutants completely defective in synthesis of cyclopropane fatty acids. J. Bacteriol. 1986, 166, 872–877. [Google Scholar] [CrossRef]
- Harley, J.B.; Santangelo, G.M.; Rasmussen, H.; Goldfine, H. Dependence of Escherichia coli hyperbaric oxygen toxicity on the lipid acyl chain composition. J. Bacteriol. 1978, 134, 808–820. [Google Scholar] [CrossRef]
- Dufourc, E.J.; Smith, I.C.; Jarrell, H.C. A 2H-NMR analysis of dihydrosterculoyl-containing lipids in model membranes: Structural effects of a cyclopropane ring. Chem. Phys. Lipids 1983, 33, 153–177. [Google Scholar] [CrossRef] [PubMed]
- Choi, T.R.; Park, Y.L.; Song, H.S.; Lee, S.M.; Park, S.L.; Lee, H.S.; Kim, H.J.; Bhatia, S.K.; Gurav, R.; Lee, Y.K.; et al. Effects of a Δ-9-fatty acid desaturase and a cyclopropane-fatty acid synthase from the novel psychrophile Pseudomonas sp. B14-6 on bacterial membrane properties. J. Ind. Microbiol. Biotechnol. 2020, 47, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Choi, T.R.; Song, H.S.; Han, Y.H.; Park, Y.L.; Park, J.Y.; Yang, S.Y.; Bhatia, S.K.; Gurav, R.; Kim, H.J.; Lee, Y.K.; et al. Enhanced tolerance to inhibitors of Escherichia coli by heterologous expression of cyclopropane-fatty acid-acyl-phospholipid synthase (cfa) from Halomonas socia. Bioprocess Biosyst. Eng. 2020, 43, 909–918. [Google Scholar] [CrossRef]
- Wang, A.Y.; Grogan, D.W.; Cronan, J.E., Jr. Cyclopropane fatty acid synthase of Escherichia coli: Deduced amino acid sequence, purification, and studies of the enzyme active site. Biochemistry 1992, 31, 11020–11028. [Google Scholar] [CrossRef]
- Barkan, D.; Liu, Z.; Sacchettini, J.C.; Glickman, M.S. Mycolic acid cyclopropanation is essential for viability, drug resistance, and cell wall integrity of Mycobacterium tuberculosis. Chem. Biol. 2009, 16, 499–509. [Google Scholar] [CrossRef]
- Barkan, D.; Hedhli, D.; Yan, H.G.; Huygen, K.; Glickman, M.S. Mycobacterium tuberculosis lacking all mycolic acid cyclopropanation is viable but highly attenuated and hyperinflammatory in mice. Infect. Immun. 2012, 80, 1958–1968. [Google Scholar] [CrossRef]
- Blunsom, N.J.; Cockcroft, S. CDP-Diacylglycerol Synthases (CDS): Gateway to Phosphatidylinositol and Cardiolipin Synthesis. Front. Cell Dev. Biol. 2020, 8, 63. [Google Scholar] [CrossRef]
- Jennings, W.; Epand, R.M. CDP-diacylglycerol, a critical intermediate in lipid metabolism. Chem. Phys. Lipids 2020, 230, 104914. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.H.; Guan, Z.; Zhao, J.; Raetz, C.R. Three phosphatidylglycerol-phosphate phosphatases in the inner membrane of Escherichia coli. J. Biol. Chem. 2011, 286, 5506–5518. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.K.; Bogdanov, M.; Zhao, J.; Dowhan, W.; Raetz, C.R.; Guan, Z. Discovery of a cardiolipin synthase utilizing phosphatidylethanolamine and phosphatidylglycerol as substrates. Proc. Natl. Acad. Sci. USA 2012, 109, 16504–16509. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Tropp, B.E. A second Escherichia coli protein with CL synthase activity. Biochim. Biophys. Acta 2000, 1483, 263–274. [Google Scholar] [CrossRef]
- Li, C.; Tan, B.K.; Zhao, J.; Guan, Z. In Vivo and In vitro Synthesis of Phosphatidylglycerol by an Escherichia coli Cardiolipin Synthase. J. Biol. Chem. 2016, 291, 25144–25153. [Google Scholar] [CrossRef]
- Tsai, M.; Ohniwa, R.L.; Kato, Y.; Takeshita, S.L.; Ohta, T.; Saito, S.; Hayashi, H.; Morikawa, K. Staphylococcus aureus requires cardiolipin for survival under conditions of high salinity. BMC Microbiol. 2011, 11, 13. [Google Scholar] [CrossRef]
- Kawai, F.; Shoda, M.; Harashima, R.; Sadaie, Y.; Hara, H.; Matsumoto, K. Cardiolipin domains in Bacillus subtilis marburg membranes. J. Bacteriol. 2004, 186, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Sohlenkamp, C.; de Rudder, K.E.; Geiger, O. Phosphatidylethanolamine is not essential for growth of Sinorhizobium meliloti on complex culture media. J. Bacteriol. 2004, 186, 1667–1677. [Google Scholar] [CrossRef]
- Sohlenkamp, C.; López-Lara, I.M.; Geiger, O. Biosynthesis of phosphatidylcholine in bacteria. Prog. Lipid Res. 2003, 42, 115–162. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Crick, D.C.; Brennan, P.J. Phosphatidylinositol is an essential phospholipid of mycobacteria. J. Biol. Chem. 2000, 275, 30092–30099. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.; Lonsdale, J.T.; Besra, G.S.; Brennan, P.J. Phosphatidylinositol synthesis in mycobacteria. Biochim. Biophys. Acta 1999, 1436, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Morii, H.; Okauchi, T.; Nomiya, H.; Ogawa, M.; Fukuda, K.; Taniguchi, H. Studies of inositol 1-phosphate analogues as inhibitors of the phosphatidylinositol phosphate synthase in mycobacteria. J. Biochem. 2013, 153, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Roy, H.; Dare, K.; Ibba, M. Adaptation of the bacterial membrane to changing environments using aminoacylated phospholipids. Mol. Microbiol. 2009, 71, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Andrä, J.; Goldmann, T.; Ernst, C.M.; Peschel, A.; Gutsmann, T. Multiple peptide resistance factor (MprF)-mediated Resistance of Staphylococcus aureus against antimicrobial peptides coincides with a modulated peptide interaction with artificial membranes comprising lysyl-phosphatidylglycerol. J. Biol. Chem. 2011, 286, 18692–18700. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.M.; Peschel, A. Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids. Mol. Microbiol. 2011, 80, 290–299. [Google Scholar] [CrossRef]
- Slavetinsky, C.J.; Hauser, J.N.; Gekeler, C.; Slavetinsky, J.; Geyer, A.; Kraus, A.; Heilingbrunner, D.; Wagner, S.; Tesar, M.; Krismer, B.; et al. Sensitizing Staphylococcus aureus to antibacterial agents by decoding and blocking the lipid flippase MprF. Elife 2022, 11, e66376. [Google Scholar] [CrossRef]
- Dhankhar, P.; Dalal, V.; Kotra, D.G.; Kumar, P. In-silico approach to identify novel potent inhibitors against GraR of S. aureus. Front. Biosci. Landmark Ed. 2020, 25, 1337–1360. [Google Scholar] [CrossRef]
- Van Horn, W.D.; Sanders, C.R. Prokaryotic diacylglycerol kinase and undecaprenol kinase. Annu. Rev. Biophys. 2012, 41, 81–101. [Google Scholar] [CrossRef]
- Baker, B.R.; Ives, C.M.; Bray, A.; Caffrey, M.; Cochrane, S.A. Undecaprenol kinase: Function, mechanism and substrate specificity of a potential antibiotic target. Eur. J. Med. Chem. 2021, 210, 113062. [Google Scholar] [CrossRef]
- Yeo, W.S.; Jeong, B.; Ullah, N.; Shah, M.A.; Ali, A.; Kim, K.K.; Bae, T. Ftsh Sensitizes Methicillin-Resistant Staphylococcus aureus to β-Lactam Antibiotics by Degrading YpfP, a Lipoteichoic Acid Synthesis Enzyme. Antibiotics 2021, 10, 1198. [Google Scholar] [CrossRef] [PubMed]
- Galloway, S.M.; Raetz, C.R. A mutant of Escherichia coli defective in the first step of endotoxin biosynthesis. J. Biol. Chem. 1990, 265, 6394–6402. [Google Scholar] [CrossRef]
- Kelly, T.M.; Stachula, S.A.; Raetz, C.R.; Anderson, M.S. The firA gene of Escherichia coli encodes UDP-3-O-(R-3-hydroxymyristoyl)-glucosamine N-acyltransferase. The third step of endotoxin biosynthesis. J. Biol. Chem. 1993, 268, 19866–19874. [Google Scholar] [CrossRef]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Immormino, R.M.; Gewirth, D.T.; Raetz, C.R. Structure of UDP-N-acetylglucosamine acyltransferase with a bound antibacterial pentadecapeptide. Proc. Natl. Acad. Sci. USA 2006, 103, 10877–10882. [Google Scholar] [CrossRef]
- Jenkins, R.J.; Dotson, G.D. Dual targeting antibacterial peptide inhibitor of early lipid A biosynthesis. ACS Chem. Biol. 2012, 7, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Ma, X.; Balibar, C.J.; Baxter Rath, C.M.; Benton, B.; Bermingham, A.; Casey, F.; Chie-Leon, B.; Cho, M.K.; Frank, A.O.; et al. Two Distinct Mechanisms of Inhibition of LpxA Acyltransferase Essential for Lipopolysaccharide Biosynthesis. J. Am. Chem. Soc. 2020, 142, 4445–4455. [Google Scholar] [CrossRef]
- Clements, J.M.; Coignard, F.; Johnson, I.; Chandler, S.; Palan, S.; Waller, A.; Wijkmans, J.; Hunter, M.G. Antibacterial activities and characterization of novel inhibitors of LpxC. Antimicrob. Agents Chemother. 2002, 46, 1793–1799. [Google Scholar] [CrossRef]
- Mdluli, K.E.; Witte, P.R.; Kline, T.; Barb, A.W.; Erwin, A.L.; Mansfield, B.E.; McClerren, A.L.; Pirrung, M.C.; Tumey, L.N.; Warrener, P.; et al. Molecular validation of LpxC as an antibacterial drug target in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2006, 50, 2178–2184. [Google Scholar] [CrossRef]
- Chen, M.H.; Steiner, M.G.; de Laszlo, S.E.; Patchett, A.A.; Anderson, M.S.; Hyland, S.A.; Onishi, H.R.; Silver, L.L.; Raetz, C.R. Carbohydroxamido-oxazolidines: Antibacterial agents that target lipid A biosynthesis. Bioorg. Med. Chem. Lett. 1999, 9, 313–318. [Google Scholar] [CrossRef]
- McClerren, A.L.; Endsley, S.; Bowman, J.L.; Andersen, N.H.; Guan, Z.; Rudolph, J.; Raetz, C.R. A slow, tight-binding inhibitor of the zinc-dependent deacetylase LpxC of lipid A biosynthesis with antibiotic activity comparable to ciprofloxacin. Biochemistry 2005, 44, 16574–16583. [Google Scholar] [CrossRef]
- Barb, A.W.; McClerren, A.L.; Snehelatha, K.; Reynolds, C.M.; Zhou, P.; Raetz, C.R. Inhibition of lipid A biosynthesis as the primary mechanism of CHIR-090 antibiotic activity in Escherichia coli. Biochemistry 2007, 46, 3793–3802. [Google Scholar] [CrossRef] [PubMed]
- Tomaras, A.P.; McPherson, C.J.; Kuhn, M.; Carifa, A.; Mullins, L.; George, D.; Desbonnet, C.; Eidem, T.M.; Montgomery, J.I.; Brown, M.F.; et al. LpxC inhibitors as new antibacterial agents and tools for studying regulation of lipid A biosynthesis in Gram-negative pathogens. mBio 2014, 5, e01551-14. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.E.; Fierke, C.A.; Tumey, L.N.; Pirrung, M.; Uchiyama, T.; Tahir, S.H.; Hindsgaul, O.; Raetz, C.R. Antibacterial agents that target lipid A biosynthesis in gram-negative bacteria. Inhibition of diverse UDP-3-O-(r-3-hydroxymyristoyl)-n-acetylglucosamine deacetylases by substrate analogs containing zinc binding motifs. J. Biol. Chem. 2000, 275, 11002–11009. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Prathapam, R.; Wartchow, C.; Chie-Leon, B.; Ho, C.M.; De Vicente, J.; Han, W.; Li, M.; Lu, Y.; Ramurthy, S.; et al. Structural and Biological Basis of Small Molecule Inhibition of Escherichia coli LpxD Acyltransferase Essential for Lipopolysaccharide Biosynthesis. ACS Infect. Dis. 2020, 6, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Bohl, H.O.; Ieong, P.; Lee, J.K.; Lee, T.; Kankanala, J.; Shi, K.; Demir, Ö.; Kurahashi, K.; Amaro, R.E.; Wang, Z.; et al. The substrate-binding cap of the UDP-diacylglucosamine pyrophosphatase LpxH is highly flexible, enabling facile substrate binding and product release. J. Biol. Chem. 2018, 293, 7969–7981. [Google Scholar] [CrossRef]
- Cho, J.; Lee, M.; Cochrane, C.S.; Webster, C.G.; Fenton, B.A.; Zhao, J.; Hong, J.; Zhou, P. Structural basis of the UDP-diacylglucosamine pyrophosphohydrolase LpxH inhibition by sulfonyl piperazine antibiotics. Proc. Natl. Acad. Sci. USA 2020, 117, 4109–4116. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Zhao, J.; Kwak, S.H.; Cho, J.; Lee, M.; Gillespie, R.A.; Kwon, D.Y.; Lee, H.; Park, H.J.; Wu, Q.; et al. Structure-Activity Relationship of Sulfonyl Piperazine LpxH Inhibitors Analyzed by an LpxE-Coupled Malachite Green Assay. ACS Infect. Dis. 2019, 5, 641–651. [Google Scholar] [CrossRef]
- Raetz, C.R.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 2007, 76, 295–329. [Google Scholar] [CrossRef]
- Williams, A.H.; Raetz, C.R. Structural basis for the acyl chain selectivity and mechanism of UDP-N-acetylglucosamine acyltransferase. Proc. Natl. Acad. Sci. USA 2007, 104, 13543–13550. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.J.; Heslip, K.A.; Meagher, J.L.; Stuckey, J.A.; Dotson, G.D. Structural basis for the recognition of peptide RJPXD33 by acyltransferases in lipid A biosynthesis. J. Biol. Chem. 2014, 289, 15527–15535. [Google Scholar] [CrossRef] [PubMed]
- Dangkulwanich, M.; Raetz, C.R.H.; Williams, A.H. Structure guided design of an antibacterial peptide that targets UDP-N-acetylglucosamine acyltransferase. Sci. Rep. 2019, 9, 3947. [Google Scholar] [CrossRef] [PubMed]
- Kroeck, K.G.; Sacco, M.D.; Smith, E.W.; Zhang, X.; Shoun, D.; Akhtar, A.; Darch, S.E.; Cohen, F.; Andrews, L.D.; Knox, J.E.; et al. Discovery of dual-activity small-molecule ligands of Pseudomonas aeruginosa LpxA and LpxD using SPR and X-ray crystallography. Sci. Rep. 2019, 9, 15450. [Google Scholar] [CrossRef]
- Bhaskar, B.V.; Babu, T.M.C.; Rammohan, A.; Zheng, G.Y.; Zyryanov, G.V.; Gu, W. Structure-Based Virtual Screening of Pseudomonas aeruginosa LpxA Inhibitors Using Pharmacophore-Based Approach. Biomolecules 2020, 10, 266. [Google Scholar] [CrossRef]
- Pratap, S.; Kesari, P.; Yadav, R.; Dev, A.; Narwal, M.; Kumar, P. Acyl chain preference and inhibitor identification of Moraxella catarrhalis LpxA: Insight through crystal structure and computational studies. Int. J. Biol. Macromol. 2017, 96, 759–765. [Google Scholar] [CrossRef]
- Shapiro, A.B.; Ross, P.L.; Gao, N.; Livchak, S.; Kern, G.; Yang, W.; Andrews, B.; Thresher, J. A high-throughput-compatible fluorescence anisotropy-based assay for competitive inhibitors of Escherichia coli UDP-N-acetylglucosamine acyltransferase (LpxA). J. Biomol. Screen. 2013, 18, 341–347. [Google Scholar] [CrossRef]
- Mochalkin, I.; Knafels, J.D.; Lightle, S. Crystal structure of LpxC from Pseudomonas aeruginosa complexed with the potent BB-78485 inhibitor. Protein Sci. 2008, 17, 450–457. [Google Scholar] [CrossRef]
- Onishi, H.R.; Pelak, B.A.; Gerckens, L.S.; Silver, L.L.; Kahan, F.M.; Chen, M.H.; Patchett, A.A.; Galloway, S.M.; Hyland, S.A.; Anderson, M.S.; et al. Antibacterial agents that inhibit lipid A biosynthesis. Science 1996, 274, 980–982. [Google Scholar] [CrossRef]
- Coggins, B.E.; McClerren, A.L.; Jiang, L.; Li, X.; Rudolph, J.; Hindsgaul, O.; Raetz, C.R.; Zhou, P. Refined solution structure of the LpxC-TU-514 complex and pKa analysis of an active site histidine: Insights into the mechanism and inhibitor design. Biochemistry 2005, 44, 1114–1126. [Google Scholar] [CrossRef]
- Caughlan, R.E.; Jones, A.K.; Delucia, A.M.; Woods, A.L.; Xie, L.; Ma, B.; Barnes, S.W.; Walker, J.R.; Sprague, E.R.; Yang, X.; et al. Mechanisms decreasing In vitro susceptibility to the LpxC inhibitor CHIR-090 in the gram-negative pathogen Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2012, 56, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Liang, X.; Chen, X.; Zeng, D.; Joo, S.H.; Chung, H.S.; Barb, A.W.; Swanson, S.M.; Nicholas, R.A.; Li, Y.; et al. Species-specific and inhibitor-dependent conformations of LpxC: Implications for antibiotic design. Chem. Biol. 2011, 18, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Lee, C.J.; Chen, X.; Chung, H.S.; Zeng, D.; Raetz, C.R.; Li, Y.; Zhou, P.; Toone, E.J. Syntheses, structures and antibiotic activities of LpxC inhibitors based on the diacetylene scaffold. Bioorg. Med. Chem. 2011, 19, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Zhao, J.; Chung, H.S.; Guan, Z.; Raetz, C.R.; Zhou, P. Mutants resistant to LpxC inhibitors by rebalancing cellular homeostasis. J. Biol. Chem. 2013, 288, 5475–5486. [Google Scholar] [CrossRef]
- Jones, A.K.; Caughlan, R.E.; Woods, A.L.; Uehara, K.; Xie, L.; Barnes, S.W.; Walker, J.R.; Thompson, K.V.; Ranjitkar, S.; Lee, P.S.; et al. Mutations Reducing In vitro Susceptibility to Novel LpxC Inhibitors in Pseudomonas aeruginosa and Interplay of Efflux and Nonefflux Mechanisms. Antimicrob. Agents Chemother. 2019, 64, e01490-19. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Lei, P.; Wang, Y.; Wang, J.; Yang, J.; Zhang, J. Small molecule LpxC inhibitors against gram-negative bacteria: Advances and future perspectives. Eur. J. Med. Chem. 2023, 253, 115326. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.N.; Ray, M.; Pattnaik, A.; Pradhan, S.K. Drug Target Identification and Elucidation of Natural Inhibitors for Bordetella petrii: An In Silico Study. Genom. Inform. 2016, 14, 241–254. [Google Scholar] [CrossRef]
- Metzger, L.E., 4th; Lee, J.K.; Finer-Moore, J.S.; Raetz, C.R.; Stroud, R.M. LpxI structures reveal how a lipid A precursor is synthesized. Nat. Struct. Mol. Biol. 2012, 19, 1132–1138. [Google Scholar] [CrossRef]
- Metzger, L.E., 4th; Raetz, C.R. An alternative route for UDP-diacylglucosamine hydrolysis in bacterial lipid A biosynthesis. Biochemistry 2010, 49, 6715–6726. [Google Scholar] [CrossRef]
- Young, H.E.; Zhao, J.; Barker, J.R.; Guan, Z.; Valdivia, R.H.; Zhou, P. Discovery of the Elusive UDP-Diacylglucosamine Hydrolase in the Lipid A Biosynthetic Pathway in Chlamydia trachomatis. mBio 2016, 7, e00090. [Google Scholar] [CrossRef]
- Nayar, A.S.; Dougherty, T.J.; Ferguson, K.E.; Granger, B.A.; McWilliams, L.; Stacey, C.; Leach, L.J.; Narita, S.; Tokuda, H.; Miller, A.A.; et al. Novel antibacterial targets and compounds revealed by a high-throughput cell wall reporter assay. J. Bacteriol. 2015, 197, 1726–1734. [Google Scholar] [CrossRef]
- Kwak, S.H.; Cochrane, C.S.; Ennis, A.F.; Lim, W.Y.; Webster, C.G.; Cho, J.; Fenton, B.A.; Zhou, P.; Hong, J. Synthesis and evaluation of sulfonyl piperazine LpxH inhibitors. Bioorg. Chem. 2020, 102, 104055. [Google Scholar] [CrossRef]
- Zhou, P.; Hong, J. Structure- and Ligand-Dynamics-Based Design of Novel Antibiotics Targeting Lipid A Enzymes LpxC and LpxH in Gram-Negative Bacteria. Acc. Chem. Res. 2021, 54, 1623–1634. [Google Scholar] [CrossRef]
- Martínez-Guitián, M.; Vázquez-Ucha, J.C.; Álvarez-Fraga, L.; Conde-Pérez, K.; Bou, G.; Poza, M.; Beceiro, A. Antisense inhibition of lpxB gene expression in Acinetobacter baumannii by peptide-PNA conjugates and synergy with colistin. J. Antimicrob. Chemother. 2020, 75, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Damale, M.G.; Pathan, S.K.; Patil, R.B.; Sangshetti, J.N. Pharmacoinformatics approaches to identify potential hits against tetraacyldisaccharide 4′-kinase (LpxK) of Pseudomonas aeruginosa. RSC Adv. 2020, 10, 32856–32874. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Quinn, P.J.; Yan, A. Kdo2 -lipid A: Structural diversity and impact on immunopharmacology. Biol. Rev. Camb. Philos. Soc. 2015, 90, 408–427. [Google Scholar] [CrossRef]
- Emiola, A.; George, J.; Andrews, S.S. A Complete Pathway Model for Lipid A Biosynthesis in Escherichia coli. PLoS ONE 2015, 10, e0121216. [Google Scholar] [CrossRef] [PubMed]
- Hankins, J.V.; Madsen, J.A.; Giles, D.K.; Childers, B.M.; Klose, K.E.; Brodbelt, J.S.; Trent, M.S. Elucidation of a novel Vibrio cholerae lipid A secondary hydroxy-acyltransferase and its role in innate immune recognition. Mol. Microbiol. 2011, 81, 1313–1329. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y.; Makovitzky, A.; Avrahami, D. Host defense peptides and lipopeptides: Modes of action and potential candidates for the treatment of bacterial and fungal infections. Curr. Protein Pept. Sci. 2006, 7, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Radek, K.; Gallo, R. Antimicrobial peptides: Natural effectors of the innate immune system. Semin. Immunopathol. 2007, 29, 27–43. [Google Scholar] [CrossRef]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dou, X.; Song, J.; Lyu, Y.; Zhu, X.; Xu, L.; Li, W.; Shan, A. Antimicrobial peptides: Promising alternatives in the post feeding antibiotic era. Med. Res. Rev. 2019, 39, 831–859. [Google Scholar] [CrossRef]
- Ahmed, T.A.E.; Hammami, R. Recent insights into structure-function relationships of antimicrobial peptides. J. Food Biochem. 2019, 43, e12546. [Google Scholar] [CrossRef] [PubMed]
- Antonov, V.F.; Petrov, V.V.; Molnar, A.A.; Predvoditelev, D.A.; Ivanov, A.S. The appearance of single-ion channels in unmodified lipid bilayer membranes at the phase transition temperature. Nature 1980, 283, 585–586. [Google Scholar] [CrossRef]
- Finkelstein, A.; Andersen, O.S. The gramicidin A channel: A review of its permeability characteristics with special reference to the single-file aspect of transport. J. Membr. Biol. 1981, 59, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Andersen, O.S.; Koeppe, R.E., 2nd. Molecular determinants of channel function. Physiol. Rev. 1992, 72 (Suppl. S4), S89–S158. [Google Scholar] [CrossRef]
- Fringeli, U.P.; Fringeli, M. Pore formation in lipid membranes by alamethicin. Proc. Natl. Acad. Sci. USA 1979, 76, 3852–3856. [Google Scholar] [CrossRef]
- Shai, Y.; Bach, D.; Yanovsky, A. Channel formation properties of synthetic pardaxin and analogues. J. Biol. Chem. 1990, 265, 20202–20209. [Google Scholar] [CrossRef]
- Capone, R.; Mustata, M.; Jang, H.; Arce, F.T.; Nussinov, R.; Lal, R. Antimicrobial protegrin-1 forms ion channels: Molecular dynamic simulation, atomic force microscopy, and electrical conductance studies. Biophys. J. 2010, 98, 2644–2652. [Google Scholar] [CrossRef]
- Watanabe, H.; Kawano, R. Channel Current Analysis for Pore-forming Properties of an Antimicrobial Peptide, Magainin 1, Using the Droplet Contact Method. Anal. Sci. 2016, 32, 57–60. [Google Scholar] [CrossRef]
- Gallucci, E.; Meleleo, D.; Micelli, S.; Picciarelli, V. Magainin 2 channel formation in planar lipid membranes: The role of lipid polar groups and ergosterol. Eur. Biophys. J. 2003, 32, 22–32. [Google Scholar] [CrossRef]
- Mellor, I.R.; Sansom, M.S. Ion-channel properties of mastoparan, a 14-residue peptide from wasp venom, and of MP3, a 12-residue analogue. Proc. R. Soc. Lond. B Biol. Sci. 1990, 239, 383–400. [Google Scholar] [CrossRef]
- Arbuzova, A.; Schwarz, G. Pore-forming action of mastoparan peptides on liposomes: A quantitative analysis. Biochim. Biophys. Acta 1999, 1420, 139–152. [Google Scholar] [CrossRef]
- Efimova, S.S.; Schagina, L.V.; Ostroumova, O.S. Channel-forming activity of cecropins in lipid bilayers: Effect of agents modifying the membrane dipole potential. Langmuir 2014, 30, 7884–7892. [Google Scholar] [CrossRef] [PubMed]
- Efimova, S.S.; Shekunov, E.V.; Chernyshova, D.N.; Zakharova, A.A.; Ostroumova, O.S. Dependence of the channel-forming ability of lantibiotics on the lipid composition of the membranes. Biochem. Suppl. Ser. A Membr. Cell Biol. 2022, 16, 144–150. [Google Scholar] [CrossRef]
- Sheth, T.R.; Henderson, R.M.; Hladky, S.B.; Cuthbert, A.W. Ion channel formation by duramycin. Biochim. Biophys. Acta 1992, 1107, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Sansom, M.S. Alamethicin and related peptaibols—Model ion channels. Eur. Biophys. J. 1993, 22, 105–124. [Google Scholar] [CrossRef]
- Gordon, L.G.; Haydon, D.A. The unit conductance channel of alamethicin. Biochim. Biophys. Acta 1972, 255, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.; Fink, J.; Merrifield, R.B.; Mauzerall, D. Channel-forming properties of cecropins and related model compounds incorporated into planar lipid membranes. Proc. Natl. Acad. Sci. USA 1988, 85, 5072–5076. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, Y.; Mirzabekov, T.; Martin, D.W.; Lehrer, R.I.; Kagan, B.L. Membrane channel formation by antimicrobial protegrins. Biochim. Biophys. Acta 1999, 1420, 23–29. [Google Scholar] [CrossRef]
- Saint, N.; Marri, L.; Marchini, D.; Molle, G. The antibacterial peptide ceratotoxin A displays alamethicin-like behavior in lipid bilayers. Peptides 2003, 24, 1779–1784. [Google Scholar] [CrossRef]
- Mayer, S.F.; Ducrey, J.; Dupasquier, J.; Haeni, L.; Rothen-Rutishauser, B.; Yang, J.; Fennouri, A.; Mayer, M. Targeting specific membranes with an azide derivative of the pore-forming peptide ceratotoxin A. Biochim. Biophys. Acta Biomembr. 2019, 1861, 183023. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Human antimicrobial peptides and proteins. Pharmaceuticals 2014, 7, 545–594. [Google Scholar] [CrossRef]
- Henzler Wildman, K.A.; Lee, D.K.; Ramamoorthy, A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry 2003, 42, 6545–6558. [Google Scholar] [CrossRef]
- Khondker, A.; Rheinstädter, M.C. How do bacterial membranes resist polymyxin antibiotics? Commun. Biol. 2020, 3, 77. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, A.; Hagart, K.L.; Klöckner, A.; Becce, M.; Evans, L.E.; Furniss, R.C.D.; Mavridou, D.A.; Murphy, R.; Stevens, M.M.; Davies, J.C.; et al. Colistin kills bacteria by targeting lipopolysaccharide in the cytoplasmic membrane. Elife 2021, 10, e65836. [Google Scholar] [CrossRef]
- Taylor, R.; Beriashvili, D.; Taylor, S.; Palmer, M. Daptomycin Pore Formation Is Restricted by Lipid Acyl Chain Composition. ACS Infect. Dis. 2017, 3, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Beriashvili, D.; Taylor, R.; Kralt, B.; Abu Mazen, N.; Taylor, S.D.; Palmer, M. Mechanistic studies on the effect of membrane lipid acyl chain composition on daptomycin pore formation. Chem. Phys. Lipids 2018, 216, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Tyurin, A.P.; Alferova, V.A.; Paramonov, A.S.; Shuvalov, M.V.; Kudryakova, G.K.; Rogozhin, E.A.; Zherebker, A.Y.; Brylev, V.A.; Chistov, A.A.; Baranova, A.A.; et al. Gausemycins-A,B: Cyclic Lipoglycopeptides from Streptomyces sp. Angew. Chem. Int. Ed. Engl. 2021, 60, 18694–18703. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Laver, D.R. The barrel-stave model as applied to alamethicin and its analogs reevaluated. Biophys. J. 1994, 66, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, F.; Buck, B.; Lee, D.K.; Hallock, K.J.; Ramamoorthy, A.; Veglia, G. Structure and orientation of pardaxin determined by NMR experiments in model membranes. J. Biol. Chem. 2004, 279, 45815–45823. [Google Scholar] [CrossRef]
- Allende, D.; Simon, S.A.; McIntosh, T.J. Melittin-induced bilayer leakage depends on lipid material properties: Evidence for toroidal pores. Biophys. J. 2005, 88, 1828–1837. [Google Scholar] [CrossRef]
- Matsuzaki, K. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta 1998, 1376, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Murzyn, K.; Pasenkiewicz-Gierula, M. Construction of a toroidal model for the magainin pore. J. Mol. Model. 2003, 9, 217–224. [Google Scholar] [CrossRef]
- Gazit, E.; Boman, A.; Boman, H.G.; Shai, Y. Interaction of the mammalian antibacterial peptide cecropin P1 with phospholipid vesicles. Biochemistry 1995, 34, 11479–11488. [Google Scholar] [CrossRef]
- Fernandez, D.I.; Le Brun, A.P.; Whitwell, T.C.; Sani, M.A.; James, M.; Separovic, F. The antimicrobial peptide aurein 1.2 disrupts model membranes via the carpet mechanism. Phys. Chem. Chem. Phys. 2012, 14, 15739–15751. [Google Scholar] [CrossRef]
- Battista, F.; Oliva, R.; Del Vecchio, P.; Winter, R.; Petraccone, L. Insights into the Action Mechanism of the Antimicrobial Peptide Lasioglossin III. Int. J. Mol. Sci. 2021, 22, 2857. [Google Scholar] [CrossRef]
- Ma, B.; Fang, C.; Lu, L.; Wang, M.; Xue, X.; Zhou, Y.; Li, M.; Hu, Y.; Luo, X.; Hou, Z. The antimicrobial peptide thanatin disrupts the bacterial outer membrane and inactivates the NDM-1 metallo-β-lactamase. Nat. Commun. 2019, 10, 3517. [Google Scholar] [CrossRef]
- Henderson, J.M.; Waring, A.J.; Separovic, F.; Lee, K.Y.C. Antimicrobial Peptides Share a Common Interaction Driven by Membrane Line Tension Reduction. Biophys. J. 2016, 111, 2176–2189. [Google Scholar] [CrossRef]
- Efimova, S.S.; Medvedev, R.Y.; Chulkov, E.G.; Ostroumova, O.S. Regulation of the Pore-Forming Activity of Cecropin A by Local Anesthetics. Cell Tiss. Biol. 2018, 12, 331–341. [Google Scholar] [CrossRef]
- Lee, C.C.; Sun, Y.; Qian, S.; Huang, H.W. Transmembrane pores formed by human antimicrobial peptide LL-37. Biophys. J. 2011, 100, 1688–1696. [Google Scholar] [CrossRef]
- Mak, D.O.; Webb, W.W. Two classes of alamethicin transmembrane channels: Molecular models from single-channel properties. Biophys. J. 1995, 69, 2323–2336. [Google Scholar] [CrossRef]
- Fennouri, A.; Mayer, S.F.; Schroeder, T.B.H.; Mayer, M. Single channel planar lipid bilayer recordings of the melittin variant MelP5. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2051–2057. [Google Scholar] [CrossRef]
- Wiedemann, I.; Benz, R.; Sahl, H.G. Lipid II-mediated pore formation by the peptide antibiotic nisin: A black lipid membrane study. J. Bacteriol. 2004, 186, 3259–3261. [Google Scholar] [CrossRef]
- Kagan, B.L.; Selsted, M.E.; Ganz, T.; Lehrer, R.I. Antimicrobial defensin peptides form voltage-dependent ion-permeable channels in planar lipid bilayer membranes. Proc. Natl. Acad. Sci. USA 1990, 87, 210–214. [Google Scholar] [CrossRef]
- Hristova, K.; Selsted, M.E.; White, S.H. Critical role of lipid composition in membrane permeabilization by rabbit neutrophil defensins. J. Biol. Chem. 1997, 272, 24224–24233. [Google Scholar] [CrossRef]
- Seydlová, G.; Sokol, A.; Lišková, P.; Konopásek, I.; Fišer, R. Daptomycin Pore Formation and Stoichiometry Depend on Membrane Potential of Target Membrane. Antimicrob. Agents Chemother. 2018, 63, e01589-18. [Google Scholar] [CrossRef]
- Peschel, A.; Jack, R.W.; Otto, M.; Collins, L.V.; Staubitz, P.; Nicholson, G.; Kalbacher, H.; Nieuwenhuizen, W.F.; Jung, G.; Tarkowski, A.; et al. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J. Exp. Med. 2001, 193, 1067–1076. [Google Scholar] [CrossRef]
- Dorrer, E.; Teuber, M. Induction of polymyxin resistance in Pseudomonas fluorescens by phosphate limitation. Arch. Microbiol. 1977, 114, 87–89. [Google Scholar] [CrossRef]
- Breazeale, S.D.; Ribeiro, A.A.; McClerren, A.L.; Raetz, C.R. A formyltransferase required for polymyxin resistance in Escherichia coli and the modification of lipid A with 4-Amino-4-deoxy-L-arabinose. Identification and function oF UDP-4-deoxy-4-formamido-L-arabinose. J. Biol. Chem. 2005, 280, 14154–14167. [Google Scholar] [CrossRef]
- Ernst, R.K.; Guina, T.; Miller, S.I. Salmonella typhimurium outer membrane remodeling: Role in resistance to host innate immunity. Microbes Infect. 2001, 3, 1327–1334. [Google Scholar] [CrossRef]
- Gunn, J.S.; Lim, K.B.; Krueger, J.; Kim, K.; Guo, L.; Hackett, M.; Miller, S.I. PmrA-PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol. Microbiol. 1998, 27, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, S.M.; Ernst, R.K.; Miller, S.I. PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid A. J. Bacteriol. 2004, 186, 575–579. [Google Scholar] [CrossRef]
- Trent, M.S.; Ribeiro, A.A.; Lin, S.; Cotter, R.J.; Raetz, C.R. An inner membrane enzyme in Salmonella and Escherichia coli that transfers 4-amino-4-deoxy-L-arabinose to lipid A: Induction on polymyxin-resistant mutants and role of a novel lipid-linked donor. J. Biol. Chem. 2001, 276, 43122–43131. [Google Scholar] [CrossRef]
- Müller, A.; Wenzel, M.; Strahl, H.; Grein, F.; Saaki, T.N.V.; Kohl, B.; Siersma, T.; Bandow, J.E.; Sahl, H.G.; Schneider, T.; et al. Daptomycin inhibits cell envelope synthesis by interfering with fluid membrane microdomains. Proc. Natl. Acad. Sci. USA 2016, 113, E7077–E7086. [Google Scholar] [CrossRef]
- Mishra, N.N.; Tran, T.T.; Seepersaud, R.; Garcia-de-la-Maria, C.; Faull, K.; Yoon, A.; Proctor, R.; Miro, J.M.; Rybak, M.J.; Bayer, A.S.; et al. Perturbations of Phosphatidate Cytidylyltransferase (CdsA) Mediate Daptomycin Resistance in Streptococcus mitis/oralis by a Novel Mechanism. Antimicrob. Agents Chemother. 2017, 61, e02435-16. [Google Scholar] [CrossRef]
- Tran, T.T.; Panesso, D.; Gao, H.; Roh, J.H.; Munita, J.M.; Reyes, J.; Diaz, L.; Lobos, E.A.; Shamoo, Y.; Mishra, N.N.; et al. Whole-genome analysis of a daptomycin-susceptible enterococcus faecium strain and its daptomycin-resistant variant arising during therapy. Antimicrob. Agents Chemother. 2013, 57, 261–268. [Google Scholar] [CrossRef]
- Poshvina, D.V.; Dilbaryan, D.S.; Kasyanov, S.P.; Sadykova, V.S.; Lapchinskaya, O.A.; Rogozhin, E.A.; Vasilchenko, A.S. Staphylococcus aureus is able to generate resistance to novel lipoglycopeptide antibiotic gausemycin A. Front. Microbiol. 2022, 13, 963979. [Google Scholar] [CrossRef]
- Boudjemaa, R.; Cabriel, C.; Dubois-Brissonnet, F.; Bourg, N.; Dupuis, G.; Gruss, A.; Lévêque-Fort, S.; Briandet, R.; Fontaine-Aupart, M.P.; Steenkeste, K. Impact of Bacterial Membrane Fatty Acid Composition on the Failure of Daptomycin to Kill Staphylococcus aureus. Antimicrob. Agents Chemother. 2018, 62, e00023-18. [Google Scholar] [CrossRef]
- Ming, X.; Daeschel, M.A. Correlation of Cellular Phospholipid Content with Nisin Resistance of Listeria monocytogenes Scott A. J. Food Prot. 1995, 58, 416–420. [Google Scholar] [CrossRef]
- Gow, N.A.R.; Latge, J.P.; Munro, C.A. The Fungal Cell Wall: Structure, Biosynthesis, and Function. Microbiol. Spectr. 2017, 5, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Sudoh, M.; Yamazaki, T.; Masubuchi, K.; Taniguchi, M.; Shimma, N.; Arisawa, M.; Yamada-Okabe, H. Identification of a novel inhibitor specific to the fungal chitin synthase. Inhibition of chitin synthase 1 arrests the cell growth, but inhibition of chitin synthase 1 and 2 is lethal in the pathogenic fungus Candida albicans. J. Biol. Chem. 2000, 275, 32901–32905. [Google Scholar] [CrossRef]
- Garcia-Effron, G.; Park, S.; Perlin, D.S. Correlating echinocandin MIC and kinetic inhibition of fks1 mutant glucan synthases for Candida albicans: Implications for interpretive breakpoints. Antimicrob. Agents Chemother. 2009, 53, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Onishi, J.; Meinz, M.; Thompson, J.; Curotto, J.; Dreikorn, S.; Rosenbach, M.; Douglas, C.; Abruzzo, G.; Flattery, A.; Kong, L.; et al. Discovery of novel antifungal (1,3)-beta-D-glucan synthase inhibitors. Antimicrob. Agents Chemother. 2000, 44, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Healey, K.R.; Katiyar, S.K.; Raj, S.; Edlind, T.D. CRS-MIS in Candida glabrata: Sphingolipids modulate echinocandin-Fks interaction. Mol. Microbiol. 2012, 86, 303–313. [Google Scholar] [CrossRef]
- Satish, S.; Jiménez-Ortigosa, C.; Zhao, Y.; Lee, M.H.; Dolgov, E.; Krüger, T.; Park, S.; Denning, D.W.; Kniemeyer, O.; Brakhage, A.A.; et al. Stress-Induced Changes in the Lipid Microenvironment of β-(1,3)-d-Glucan Synthase Cause Clinically Important Echinocandin Resistance in Aspergillus fumigatus. mBio 2019, 10, e00779-19. [Google Scholar] [CrossRef]
- Ren, Z.; Chhetri, A.; Guan, Z.; Suo, Y.; Yokoyama, K.; Lee, S.Y. Structural basis for inhibition and regulation of a chitin synthase from Candida albicans. Nat. Struct. Mol. Biol. 2022, 29, 653–664. [Google Scholar] [CrossRef]
- Hu, X.; Yang, P.; Chai, C.; Liu, J.; Sun, H.; Wu, Y.; Zhang, M.; Zhang, M.; Liu, X.; Yu, H. Structural and mechanistic insights into fungal β-1,3-glucan synthase FKS1. Nature 2023, 616, 190–198. [Google Scholar] [CrossRef]
- Leibundgut, M.; Maier, T.; Jenni, S.; Ban, N. The multienzyme architecture of eukaryotic fatty acid synthases. Curr. Opin. Struct. Biol. 2008, 18, 714–725. [Google Scholar] [CrossRef]
- Maier, T.; Jenni, S.; Ban, N. Architecture of mammalian fatty acid synthase at 4.5 A resolution. Science 2006, 311, 1258–1262. [Google Scholar] [CrossRef]
- Jenni, S.; Leibundgut, M.; Maier, T.; Ban, N. Architecture of a fungal fatty acid synthase at 5 A resolution. Science 2006, 311, 1263–1267. [Google Scholar] [CrossRef]
- Chayakulkeeree, M.; Rude, T.H.; Toffaletti, D.L.; Perfect, J.R. Fatty acid synthesis is essential for survival of Cryptococcus neoformans and a potential fungicidal target. Antimicrob. Agents Chemother. 2007, 51, 3537–3545. [Google Scholar] [CrossRef]
- Zhao, X.J.; McElhaney-Feser, G.E.; Bowen, W.H.; Cole, M.F.; Broedel, S.E., Jr.; Cihlar, R.L. Requirement for the Candida albicans FAS2 gene for infection in a rat model of oropharyngeal candidiasis. Microbiology 1996, 142, 2509–2514. [Google Scholar] [CrossRef]
- Zhao, X.J.; McElhaney-Feser, G.E.; Sheridan, M.J.; Broedel, S.E., Jr.; Cihlar, R.L. Avirulence of Candida albicans FAS2 mutants in a mouse model of systemic candidiasis. Infect. Immun. 1997, 65, 829–832. [Google Scholar] [CrossRef]
- Faergeman, N.J.; Black, P.N.; Zhao, X.D.; Knudsen, J.; DiRusso, C.C. The Acyl-CoA synthetases encoded within FAA1 and FAA4 in Saccharomyces cerevisiae function as components of the fatty acid transport system linking import, activation, and intracellular Utilization. J. Biol. Chem. 2001, 276, 37051–37059. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Wiltschi, B.; Kumari, P.; Kessler, B.; Vonrhein, C.; Vonck, J.; Oesterhelt, D.; Grininger, M. Inhibition of the fungal fatty acid synthase type I multienzyme complex. Proc. Natl. Acad. Sci. USA 2008, 105, 12803–12808. [Google Scholar] [CrossRef]
- DeJarnette, C.; Meyer, C.J.; Jenner, A.R.; Butts, A.; Peters, T.; Cheramie, M.N.; Phelps, G.A.; Vita, N.A.; Loudon-Hossler, V.C.; Lee, R.E.; et al. Identification of Inhibitors of Fungal Fatty Acid Biosynthesis. ACS Infect. Dis. 2021, 7, 3210–3223. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Sillaots, S.; Davison, J.; Hu, W.; Jiang, B.; Kauffman, S.; Martel, N.; Ocampo, P.; Oh, C.; Trosok, S.; et al. Chemical genetic profiling and characterization of small-molecule compounds that affect the biosynthesis of unsaturated fatty acids in Candida albicans. J. Biol. Chem. 2009, 284, 19754–19764. [Google Scholar] [CrossRef] [PubMed]
- Sant, D.G.; Tupe, S.G.; Ramana, C.V.; Deshpande, M.V. Fungal cell membrane-promising drug target for antifungal therapy. J. Appl. Microbiol. 2016, 121, 1498–1510. [Google Scholar] [CrossRef] [PubMed]
- Carman, G.M.; Han, G.S. Regulation of phospholipid synthesis in the yeast Saccharomyces cerevisiae. Annu. Rev. Biochem. 2011, 80, 859–883. [Google Scholar] [CrossRef]
- Pan, J.; Hu, C.; Yu, J.H. Lipid Biosynthesis as an Antifungal Target. J. Fungi 2018, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- McDonough, V.M.; Buxeda, R.J.; Bruno, M.E.; Ozier-Kalogeropoulos, O.; Adeline, M.T.; McMaster, C.R.; Bell, R.M.; Carman, G.M. Regulation of phospholipid biosynthesis in Saccharomyces cerevisiae by CTP. J. Biol. Chem. 1995, 270, 18774–18780. [Google Scholar] [CrossRef] [PubMed]
- Tams, R.N.; Cassilly, C.D.; Anaokar, S.; Brewer, W.T.; Dinsmore, J.T.; Chen, Y.L.; Patton-Vogt, J.; Reynolds, T.B. Overproduction of Phospholipids by the Kennedy Pathway Leads to Hypervirulence in Candida albicans. Front. Microbiol. 2019, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Oh, J.Y.; Hwang, B.K.; Kim, K.D. Variation in sensitivity of Magnaporthe oryzae isolates from Korea to edifenphos and iprobenfos. Crop Prot. 2008, 27, 1464–1470. [Google Scholar] [CrossRef]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef]
- Kennedy, E.P. Metabolism of lipides. Annu. Rev. Biochem. 1957, 26, 119–148. [Google Scholar] [CrossRef]
- Nagiec, M.M.; Nagiec, E.E.; Baltisberger, J.A.; Wells, G.B.; Lester, R.L.; Dickson, R.C. Sphingolipid synthesis as a target for antifungal drugs. Complementation of the inositol phosphorylceramide synthase defect in a mutant strain of Saccharomyces cerevisiae by the AUR1 gene. J. Biol. Chem. 1997, 272, 9809–9817. [Google Scholar] [CrossRef]
- Dickson, R.C. Roles for sphingolipids in Saccharomyces cerevisiae. Adv. Exp. Med. Biol. 2010, 688, 217–231. [Google Scholar] [CrossRef]
- Obeid, L.M.; Okamoto, Y.; Mao, C. Yeast sphingolipids: Metabolism and biology. Biochim. Biophys. Acta 2002, 1585, 163–171. [Google Scholar] [CrossRef]
- Ren, J.; Snider, J.; Airola, M.V.; Zhong, A.; Rana, N.A.; Obeid, L.M.; Hannun, Y.A. Quantification of 3-ketodihydrosphingosine using HPLC-ESI-MS/MS to study SPT activity in yeast Saccharomyces cerevisiae. J. Lipid Res. 2018, 59, 162–170. [Google Scholar] [CrossRef]
- Dickson, R.C.; Lester, R.L. Sphingolipid functions in Saccharomyces cerevisiae. Biochim. Biophys. Acta 2002, 1583, 13–25. [Google Scholar] [CrossRef]
- Dickson, R.C.; Sumanasekera, C.; Lester, R.L. Functions and metabolism of sphingolipids in Saccharomyces cerevisiae. Prog. Lipid Res. 2006, 45, 447–465. [Google Scholar] [CrossRef]
- Whaley, H.A. The structure of lipoxamycin, a novel antifungal antibiotic. J. Am. Chem. Soc. 1971, 93, 3767–3769. [Google Scholar] [CrossRef]
- Mandala, S.M.; Frommer, B.R.; Thornton, R.A.; Kurtz, M.B.; Young, N.M.; Cabello, M.A.; Genilloud, O.; Liesch, J.M.; Smith, J.L.; Horn, W.S. Inhibition of serine palmitoyl-transferase activity by lipoxamycin. J. Antibiot. 1994, 47, 376–379. [Google Scholar] [CrossRef]
- Kluepfel, D.; Bagli, J.; Baker, H.; Charest, M.P.; Kudelski, A. Myriocin, a new antifungal antibiotic from Myriococcum albomyces. J. Antibiot. 1972, 25, 109–115. [Google Scholar] [CrossRef] [PubMed]
- VanMiddlesworth, F.; Giacobbe, R.A.; Lopez, M.; Garrity, G.; Bland, J.A.; Bartizal, K.; Fromtling, R.A.; Polishook, J.; Zweerink, M.; Edison, A.M.; et al. Sphingofungins A, B, C, and D; a new family of antifungal agents. I. Fermentation, isolation, and biological activity. J. Antibiot. 1992, 45, 861–867. [Google Scholar] [CrossRef]
- Mandala, S.M.; Thornton, R.A.; Frommer, B.R.; Curotto, J.E.; Rozdilsky, W.; Kurtz, M.B.; Giacobbe, R.A.; Bills, G.F.; Cabello, M.A.; Martín, I.; et al. The discovery of australifungin, a novel inhibitor of sphinganine N-acyltransferase from Sporormiella australis. Producing organism, fermentation, isolation, and biological activity. J. Antibiot. 1995, 48, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Raguž, L.; Peng, C.C.; Kaiser, M.; Görls, H.; Beemelmanns, C. A Modular Approach to the Antifungal Sphingofungin Family: Concise Total Synthesis of Sphingofungin A and C. Angew. Chem. Int. Ed. Engl. 2022, 61, e202112616. [Google Scholar] [CrossRef] [PubMed]
- Mandala, S.M.; Thornton, R.A.; Frommer, B.R.; Dreikorn, S.; Kurtz, M.B. Viridiofungins, novel inhibitors of sphingolipid synthesis. J. Antibiot. 1997, 50, 339–343. [Google Scholar] [CrossRef]
- Delgado, A.; Casas, J.; Llebaria, A.; Abad, J.L.; Fabrias, G. Inhibitors of sphingolipid metabolism enzymes. Biochim. Biophys. Acta 2006, 1758, 1957–1977. [Google Scholar] [CrossRef]
- Gable, K.; Slife, H.; Bacikova, D.; Monaghan, E.; Dunn, T.M. Tsc3p is an 80-amino acid protein associated with serine palmitoyltransferase and required for optimal enzyme activity. J. Biol. Chem. 2000, 275, 7597–7603. [Google Scholar] [CrossRef]
- Yoo, H.S.; Norred, W.P.; Wang, E.; Merrill, A.H., Jr.; Riley, R.T. Fumonisin inhibition of de novo sphingolipid biosynthesis and cytotoxicity are correlated in LLC-PK1 cells. Toxicol. Appl. Pharmacol. 1992, 114, 9–15. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, K.; Normile, T.G.; Del Poeta, M. Antifungal Drug Development: Targeting the Fungal Sphingolipid Pathway. J. Fungi 2020, 6, 142. [Google Scholar] [CrossRef] [PubMed]
- Heidler, S.A.; Radding, J.A. The AUR1 gene in Saccharomyces cerevisiae encodes dominant resistance to the antifungal agent aureobasidin A (LY295337). Antimicrob. Agents Chemother. 1995, 39, 2765–2769. [Google Scholar] [CrossRef]
- Mandala, S.M.; Thornton, R.A.; Rosenbach, M.; Milligan, J.; Garcia-Calvo, M.; Bull, H.G.; Kurtz, M.B. Khafrefungin, a novel inhibitor of sphingolipid synthesis. J. Biol. Chem. 1997, 272, 32709–32714. [Google Scholar] [CrossRef]
- Ohnuki, T.; Yano, T.; Ono, Y.; Kozuma, S.; Suzuki, T.; Ogawa, Y.; Takatsu, T. Haplofungins, novel inositol phosphorylceramide synthase inhibitors, from Lauriomyces bellulus SANK 26899 I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. 2009, 62, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Aoyagi, A.; Kozuma, S.; Kawamura, Y.; Tanaka, I.; Suzuki, Y.; Takamatsu, Y.; Takatsu, T.; Inukai, M. Pleofungins, novel inositol phosphorylceramide synthase inhibitors, from Phoma sp. SANK 13899. I. Taxonomy, fermentation, isolation, and biological activities. J. Antibiot. 2007, 60, 136–142. [Google Scholar] [CrossRef]
- Mandala, S.M.; Thornton, R.A.; Milligan, J.; Rosenbach, M.; Garcia-Calvo, M.; Bull, H.G.; Harris, G.; Abruzzo, G.K.; Flattery, A.M.; Gill, C.J.; et al. Rustmicin, a potent antifungal agent, inhibits sphingolipid synthesis at inositol phosphoceramide synthase. J. Biol. Chem. 1998, 273, 14942–14949. [Google Scholar] [CrossRef]
- Harris, G.H.; Shafiee, A.; Cabello, M.A.; Curotto, J.E.; Genilloud, O.; Göklen, K.E.; Kurtz, M.B.; Rosenbach, M.; Salmon, P.M.; Thornton, R.A.; et al. Inhibition of fungal sphingolipid biosynthesis by rustmicin, galbonolide B and their new 21-hydroxy analogs. J. Antibiot. 1998, 51, 837–844. [Google Scholar] [CrossRef]
- Fauth, U.; Zähner, H.; Mühlenfeld, A.; Achenbach, H. Galbonolides A and B--two non-glycosidic antifungal macrolides. J. Antibiot. 1986, 39, 1760–1764. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Mori, Y.; Okuyama, K.; Tanikawa, K.; Yasuda, S.; Hanada, K.; Kobayashi, S. Chemistry and biology of khafrefungin. Large-scale synthesis, design, and structure-activity relationship of khafrefungin, an antifungal agent. Org. Biomol. Chem. 2003, 1, 3362–3376. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Jeffries, M.W.; Georgopapadakou, N.H. Inhibition of inositol phosphorylceramide synthase by aureobasidin A in Candida and Aspergillus species. Antimicrob. Agents Chemother. 2000, 44, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Lemetais, G.; Ferreira, T.; Cayot, P.; Gervais, P.; Beney, L. Ergosterol biosynthesis: A fungal pathway for life on land? Evolution 2012, 66, 2961–2968. [Google Scholar] [CrossRef]
- Jordá, T.; Puig, S. Regulation of Ergosterol Biosynthesis in Saccharomyces cerevisiae. Genes 2020, 11, 795. [Google Scholar] [CrossRef]
- Hu, Z.; He, B.; Ma, L.; Sun, Y.; Niu, Y.; Zeng, B. Recent Advances in Ergosterol Biosynthesis and Regulation Mechanisms in Saccharomyces cerevisiae. Indian J. Microbiol. 2017, 57, 270–277. [Google Scholar] [CrossRef]
- Miziorko, H.M. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch. Biochem. Biophys. 2011, 505, 131–143. [Google Scholar] [CrossRef]
- Tavakkoli, A.; Johnston, T.P.; Sahebkar, A. Antifungal effects of statins. Pharmacol. Ther. 2020, 208, 107483. [Google Scholar] [CrossRef]
- Ting, M.; Whitaker, E.J.; Albandar, J.M. Systematic review of the In vitro effects of statins on oral and perioral microorganisms. Eur. J. Oral Sci. 2016, 124, 4–10. [Google Scholar] [CrossRef]
- Westermeyer, C.; Macreadie, I.G. Simvastatin reduces ergosterol levels, inhibits growth and causes loss of mtDNA in Candida glabrata. FEMS Yeast Res. 2007, 7, 436–441. [Google Scholar] [CrossRef]
- Gyetvai, A.; Emri, T.; Takács, K.; Dergez, T.; Fekete, A.; Pesti, M.; Pócsi, I.; Lenkey, B. Lovastatin possesses a fungistatic effect against Candida albicans, but does not trigger apoptosis in this opportunistic human pathogen. FEMS Yeast Res. 2006, 6, 1140–1148. [Google Scholar] [CrossRef]
- Liu, G.; Vellucci, V.F.; Kyc, S.; Hostetter, M.K. Simvastatin inhibits Candida albicans biofilm In vitro. Pediatr. Res. 2009, 66, 600–604. [Google Scholar] [CrossRef]
- Rosales-Acosta, B.; Mendieta, A.; Zúñiga, C.; Tamariz, J.; Hernández Rodríguez, C.; Ibarra-García, J.A.; Villa-Tanaca, L. Simvastatin and other inhibitors of the enzyme 3-hydroxy-3-methylglutaryl coenzyme A reductase of Ustilago maydis (Um-Hmgr) affect the viability of the fungus, its synthesis of sterols and mating. Rev. Iberoam. Micol. 2019, 36, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Florin-Christensen, M.; Florin-Christensen, J.; Garin, C.; Isola, E.; Brenner, R.R.; Rasmussen, L. Inhibition of Trypanosoma cruzi growth and sterol biosynthesis by lovastatin. Biochem. Biophys. Res. Commun. 1990, 166, 1441–1445. [Google Scholar] [CrossRef] [PubMed]
- Callegari, S.; McKinnon, R.A.; Andrews, S.; de Barros Lopes, M.A. Atorvastatin-induced cell toxicity in yeast is linked to disruption of protein isoprenylation. FEMS Yeast Res. 2010, 10, 1881–1898. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Kontoyiannis, D.P.; Wan, Z.; Li, R.; Liu, W. Antifungal activity of statins against Aspergillus species. Med. Mycol. 2007, 45, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Macreadie, I.G.; Johnson, G.; Schlosser, T.; Macreadie, P.I. Growth inhibition of Candida species and Aspergillus fumigatus by statins. FEMS Microbiol. Lett. 2006, 262, 9–13. [Google Scholar] [CrossRef]
- Chin, N.X.; Weitzman, I.; Della-Latta, P. In vitro activity of fluvastatin, a cholesterol-lowering agent, and synergy with flucanazole and itraconazole against Candida species and Cryptococcus neoformans. Antimicrob. Agents Chemother. 1997, 41, 850–852. [Google Scholar] [CrossRef] [PubMed]
- Chamilos, G.; Lewis, R.E.; Kontoyiannis, D.P. Lovastatin has significant activity against zygomycetes and interacts synergistically with voriconazole. Antimicrob. Agents Chemother. 2006, 50, 96–103. [Google Scholar] [CrossRef]
- Madrigal-Aguilar, D.A.; Gonzalez-Silva, A.; Rosales-Acosta, B.; Bautista-Crescencio, C.; Ortiz-Álvarez, J.; Escalante, C.H.; Sánchez-Navarrete, J.; Hernández-Rodríguez, C.; Chamorro-Cevallos, G.; Tamariz, J.; et al. Antifungal Activity of Fibrate-Based Compounds and Substituted Pyrroles That Inhibit the Enzyme 3-Hydroxy-methyl-glutaryl-CoA Reductase of Candida glabrata (CgHMGR), Thus Decreasing Yeast Viability and Ergosterol Synthesis. Microbiol. Spectr. 2022, 10, e0164221. [Google Scholar] [CrossRef]
- Brilhante, R.S.N.; Fonseca, X.M.Q.C.; Pereira, V.S.; Araújo, G.D.S.; Oliveira, J.S.; Garcia, L.G.S.; Rodrigues, A.M.; Camargo, Z.P.; Pereira-Neto, W.A.; Castelo-Branco, D.S.C.M.; et al. In vitro inhibitory effect of statins on planktonic cells and biofilms of the Sporothrix schenckii species complex. J. Med. Microbiol. 2020, 69, 838–843. [Google Scholar] [CrossRef]
- Darwazeh, A.; Al-Shorman, H.; Mrayan, B. Effect of statin therapy on oral Candida carriage in hyperlipidemia patients: A pioneer study. Dent. Med. Probl. 2022, 59, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, A.P.; Tatara, A.M.; Shirazi, F.; Gebremariam, T.; Albert, N.D.; Lewis, R.E.; Ibrahim, A.S.; Kontoyiannis, D.P. Statin Concentrations Below the Minimum Inhibitory Concentration Attenuate the Virulence of Rhizopus oryzae. J. Infect. Dis. 2016, 214, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Tavakkoli, A.; Johnston, T.P.; Sahebkar, A. Fluvastatin: A choice for COVID-19 associated mucormycosis management. Curr. Med. Chem. 2023, 6. E-pub Ahead of Print. [Google Scholar] [CrossRef]
- Hussain, M.K.; Ahmed, S.; Khan, A.; Siddiqui, A.J.; Khatoon, S.; Jahan, S. Mucormycosis: A hidden mystery of fungal infection, possible diagnosis, treatment and development of new therapeutic agents. Eur. J. Med. Chem. 2023, 246, 115010. [Google Scholar] [CrossRef] [PubMed]
- Klug, L.; Daum, G. Yeast lipid metabolism at a glance. FEMS Yeast Res. 2014, 14, 369–388. [Google Scholar] [CrossRef]
- Plochocka, D.; Karst, F.; Swiezewska, E.; Szkopińska, A. The role of ERG20 gene (encoding yeast farnesyl diphosphate synthase) mutation in long dolichol formation. Molecular modeling of FPP synthase. Biochimie 2000, 82, 733–738. [Google Scholar] [CrossRef]
- Bergstrom, J.D.; Dufresne, C.; Bills, G.F.; Nallin-Omstead, M.; Byrne, K. Discovery, biosynthesis, and mechanism of action of the zaragozic acids: Potent inhibitors of squalene synthase. Annu. Rev. Microbiol. 1995, 49, 607–639. [Google Scholar] [CrossRef]
- Pospiech, M.; Owens, S.E.; Miller, D.J.; Austin-Muttitt, K.; Mullins, J.G.L.; Cronin, J.G.; Allemann, R.K.; Sheldon, I.M. Bisphosphonate inhibitors of squalene synthase protect cells against cholesterol-dependent cytolysins. FASEB J. 2021, 35, e21640. [Google Scholar] [CrossRef]
- Ryder, N.S. Inhibition of squalene epoxidase and sterol side-chain methylation by allylamines. Biochem. Soc. Trans. 1990, 18, 45–46. [Google Scholar] [CrossRef]
- Birnbaum, J.E. Pharmacology of the allylamines. J. Am. Acad. Dermatol. 1990, 23, 782–785. [Google Scholar] [CrossRef]
- Favre, B.; Ryder, N.S. Characterization of squalene epoxidase activity from the dermatophyte Trichophyton rubrum and its inhibition by terbinafine and other antimycotic agents. Antimicrob. Agents Chemother. 1996, 40, 443–447. [Google Scholar] [CrossRef]
- Astvad, K.M.T.; Hare, R.K.; Jørgensen, K.M.; Saunte, D.M.L.; Thomsen, P.K.; Arendrup, M.C. Increasing Terbinafine Resistance in Danish Trichophyton Isolates 2019–2020. J. Fungi 2022, 8, 150. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sabater, A.; Normand, A.C.; Bidaud, A.L.; Cremer, G.; Foulet, F.; Brun, S.; Bonnal, C.; Aït-Ammar, N.; Jabet, A.; Ayachi, A.; et al. Terbinafine Resistance in Dermatophytes: A French Multicenter Prospective Study. J. Fungi 2022, 8, 220. [Google Scholar] [CrossRef]
- Gaurav, V.; Bhattacharya, S.N.; Sharma, N.; Datt, S.; Kumar, P.; Rai, G.; Singh, P.K.; Taneja, B.; Das, S. Terbinafine resistance in dermatophytes: Time to revisit alternate antifungal therapy. J. Mycol. Med. 2021, 31, 101087. [Google Scholar] [CrossRef] [PubMed]
- Ajit, C.; Suvannasankha, A.; Zaeri, N.; Munoz, S.J. Terbinafine-associated hepatotoxicity. Am. J. Med. Sci. 2003, 325, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Trösken, E.R.; Adamska, M.; Arand, M.; Zarn, J.A.; Patten, C.; Völkel, W.; Lutz, W.K. Comparison of lanosterol-14 alpha-demethylase (CYP51) of human and Candida albicans for inhibition by different antifungal azoles. Toxicology 2006, 228, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Singh, K.; Sharma, A.; Kaur, K.; Chadha, R.; Bedi, P.M.S. Recent advances in antifungal drug development targeting lanosterol 14α-demethylase (CYP51): A comprehensive review with structural and molecular insights. Chem. Biol. Drug Des. 2023, 102, 606–639. [Google Scholar] [CrossRef]
- Sabatelli, F.; Patel, R.; Mann, P.A.; Mendrick, C.A.; Norris, C.C.; Hare, R.; Loebenberg, D.; Black, T.A.; McNicholas, P.M. In vitro activities of posaconazole, fluconazole, itraconazole, voriconazole, and amphotericin B against a large collection of clinically important molds and yeasts. Antimicrob. Agents Chemother. 2006, 50, 2009–2015. [Google Scholar] [CrossRef]
- Shafiei, M.; Peyton, L.; Hashemzadeh, M.; Foroumadi, A. History of the development of antifungal azoles: A review on structures, SAR, and mechanism of action. Bioorg. Chem. 2020, 104, 104240. [Google Scholar] [CrossRef]
- Khan, A.; Iqbal, A.; Ahmedi, S.; Manzoor, N.; Siddiqui, T. Synthesis, Anti-Fungal Potency and In silico Studies of Novel Steroidal 1,4-Dihydropyridines. Chem. Biodivers. 2023, 20, e202300096. [Google Scholar] [CrossRef]
- Pristov, K.E.; Ghannoum, M.A. Resistance of Candida to azoles and echinocandins worldwide. Clin. Microbiol. Infect. 2019, 25, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Shaw, D.; Joshi, H.; Singh, S.; Chakrabarti, A.; Rudramurthy, S.M.; Ghosh, A.K. Mechanisms of azole antifungal resistance in clinical isolates of Candida tropicalis. PLoS ONE 2022, 17, e0269721. [Google Scholar] [CrossRef]
- Husselstein, T.; Schaller, H.; Gachotte, D.; Benveniste, P. Delta7-sterol-C5-desaturase: Molecular characterization and functional expression of wild-type and mutant alleles. Plant Mol. Biol. 1999, 39, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, K.; Jones, C.W.; Stephens, C.M.; Vatsyayan, R.; Marshall, J.A.; Nes, W.D. Molecular probing of the Saccharomyces cerevisiae sterol 24-C methyltransferase reveals multiple amino acid residues involved with C2-transfer activity. Biochim. Biophys. Acta 2008, 1781, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Kaneshiro, E.S.; Johnston, L.Q.; Nkinin, S.W.; Romero, B.I.; Giner, J.L. Sterols of Saccharomyces cerevisiae erg6 Knockout Mutant Expressing the Pneumocystis carinii S-Adenosylmethionine:Sterol C-24 Methyltransferase. J. Eukaryot. Microbiol. 2015, 62, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Krauß, J.; Müller, C.; Klimt, M.; Valero, L.J.; Martínez, J.F.; Müller, M.; Bartel, K.; Binder, U.; Bracher, F. Synthesis, Biological Evaluation, and Structure-Activity Relationships of 4-Aminopiperidines as Novel Antifungal Agents Targeting Ergosterol Biosynthesis. Molecules 2021, 26, 7208. [Google Scholar] [CrossRef] [PubMed]
- Jachak, G.R.; Ramesh, R.; Sant, D.G.; Jorwekar, S.U.; Jadhav, M.R.; Tupe, S.G.; Deshpande, M.V.; Reddy, D.S. Silicon Incorporated Morpholine Antifungals: Design, Synthesis, and Biological Evaluation. ACS Med. Chem. Lett. 2015, 6, 1111–1116. [Google Scholar] [CrossRef]
- Hata, M.; Yoshida, K.; Ishii, C.; Otani, T.; Ando, A. In vitro and in vivo antifungal activities of aminopiperidine derivatives, novel ergosterol synthesis inhibitors. Biol. Pharm. Bull. 2010, 33, 473–476. [Google Scholar] [CrossRef]
- Mitsche, M.A.; McDonald, J.G.; Hobbs, H.H.; Cohen, J.C. Flux analysis of cholesterol biosynthesis in vivo reveals multiple tissue and cell-type specific pathways. Elife 2015, 4, e07999. [Google Scholar] [CrossRef]
- Benveniste, P. Sterol metabolism. Arab. Book 2002, 1, e0004. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.; Parker, J.E.; Kelly, D.E.; Kelly, S.L. Azole affinity of sterol 14α-demethylase (CYP51) enzymes from Candida albicans and Homo sapiens. Antimicrob. Agents Chemother. 2013, 57, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.; Parker, J.E.; Price, C.L.; Nes, W.D.; Garvey, E.P.; Hoekstra, W.J.; Schotzinger, R.J.; Kelly, D.E.; Kelly, S.L. The Investigational Drug VT-1129 Is a Highly Potent Inhibitor of Cryptococcus Species CYP51 but Only Weakly Inhibits the Human Enzyme. Antimicrob. Agents Chemother. 2016, 60, 4530–4538. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.; Price, C.L.; Parker, J.E.; Rolley, N.J.; Smyrniotis, C.J.; Hughes, D.D.; Thoss, V.; Nes, W.D.; Kelly, D.E.; Holman, T.R.; et al. Azole Antifungal Sensitivity of Sterol 14α-Demethylase (CYP51) and CYP5218 from Malassezia globosa. Sci. Rep. 2016, 6, 27690. [Google Scholar] [CrossRef]
- Hata, M.; Ishii, Y.; Watanabe, E.; Uoto, K.; Kobayashi, S.; Yoshida, K.; Otani, T.; Ando, A. Inhibition of ergosterol synthesis by novel antifungal compounds targeting C-14 reductase. Med. Mycol. 2010, 48, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Agner, G.; Kaulin, Y.A.; Gurnev, P.A.; Szabo, Z.; Schagina, L.V.; Takemoto, J.Y.; Blasko, K. Membrane-permeabilizing activities of cyclic lipodepsipeptides, syringopeptin 22A and syringomycin E from Pseudomonas syringae pv. syringae in human red blood cells and in bilayer lipid membranes. Bioelectrochemistry 2000, 52, 161–167. [Google Scholar]
- Hutchison, M.L.; Gross, D.C. Lipopeptide phytotoxins produced by Pseudomonas syringae pv. syringae: Comparison of the biosurfactant and ion channel-forming activities of syringopeptin and syringomycin. Mol. Plant Microbe Interact. 1997, 10, 347–354. [Google Scholar] [CrossRef]
- Bensaci, M.F.; Gurnev, P.A.; Bezrukov, S.M.; Takemoto, J.Y. Fungicidal Activities and Mechanisms of Action of Pseudomonas syringae pv. syringae Lipodepsipeptide Syringopeptins 22A and 25A. Front. Microbiol. 2011, 2, 216. [Google Scholar] [CrossRef]
- Falardeau, J.; Wise, C.; Novitsky, L.; Avis, T.J. Ecological and mechanistic insights into the direct and indirect antimicrobial properties of Bacillus subtilis lipopeptides on plant pathogens. J. Chem. Ecol. 2013, 39, 869–878. [Google Scholar] [CrossRef]
- Hutchison, M.L.; Tester, M.A.; Gross, D.C. Role of biosurfactant and ion channel-forming activities of syringomycin in transmembrane ion flux: A model for the mechanism of action in the plant-pathogen interaction. Mol. Plant Microbe Interact. 1995, 8, 610–620. [Google Scholar] [CrossRef]
- Maget-Dana, R.; Heitz, F.; Ptak, M.; Peypoux, F.; Guinand, M. Bacterial lipopeptides induce ion-conducting pores in planar bilayers. Biochem. Biophys. Res. Commun. 1985, 129, 965–971. [Google Scholar] [CrossRef]
- Malev, V.V.; Schagina, L.V.; Gurnev, P.A.; Takemoto, J.Y.; Nestorovich, E.M.; Bezrukov, S.M. Syringomycin E channel: A lipidic pore stabilized by lipopeptide? Biophys. J. 2002, 82, 1985–1994. [Google Scholar] [CrossRef]
- Ostroumova, O.S.; Gurnev, P.A.; Schagina, L.V.; Bezrukov, S.M. Asymmetry of syringomycin E channel studied by polymer partitioning. FEBS Lett. 2007, 581, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Ostroumova, O.S.; Malev, V.V.; Ilin, M.G.; Schagina, L.V. Surfactin activity depends on the membrane dipole potential. Langmuir 2010, 26, 15092–15097. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, A.A.; Efimova, S.S.; Malev, V.V.; Ostroumova, O.S. Fengycin induces ion channels in lipid bilayers mimicking target fungal cell membranes. Sci. Rep. 2019, 9, 16034. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, J.D.; Jumarie, C.; Cooper, D.G.; Laprade, R. Ionic channels induced by surfactin in planar lipid bilayer membranes. Biochim. Biophys. Acta 1991, 1064, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Maget-Dana, R.; Peypoux, F. Iturins, a special class of pore-forming lipopeptides: Biological and physicochemical properties. Toxicology 1994, 87, 151–174. [Google Scholar] [CrossRef]
- Dalla Serra, M.; Bernhart, I.; Nordera, P.; Di Giorgio, D.; Ballio, A.; Menestrina, G. Conductive properties and gating of channels formed by syringopeptin 25A, a bioactive lipodepsipeptide from Pseudomonas syringae pv. syringae, in planar lipid membranes. Mol. Plant Microbe Interact. 1999, 12, 401–409. [Google Scholar] [CrossRef]
- Maget-Dana, R.; Ptak, M.; Peypoux, F.; Michel, G. Pore-forming properties of iturin A, a lipopeptide antibiotic. Biochim. Biophys. Acta 1985, 815, 405–409. [Google Scholar] [CrossRef]
- Maget-Dana, R.; Ptak, M. Iturin lipopeptides: Interactions of mycosubtilin with lipids in planar membranes and mixed monolayers. Biochim. Biophys. Acta 1990, 1023, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Ermishkin, L.N.; Kasumov, K.M.; Potzeluyev, V.M. Single ionic channels induced in lipid bilayers by polyene antibiotics amphotericin B and nystatine. Nature 1976, 262, 698–699. [Google Scholar] [CrossRef]
- Ostroumova, O.S.; Efimova, S.S.; Schagina, L.V. Probing amphotericin B single channel activity by membrane dipole modifiers. PLoS ONE 2012, 7, e30261. [Google Scholar] [CrossRef] [PubMed]
- Tevyashova, A.; Efimova, S.; Alexandrov, A.; Omelchuk, O.; Ghazy, E.; Bychkova, E.; Zatonsky, G.; Grammatikova, N.; Dezhenkova, L.; Solovieva, S.; et al. Semisynthetic Amides of Amphotericin B and Nystatin A1: A Comparative Study of In vitro Activity/Toxicity Ratio in Relation to Selectivity to Ergosterol Membranes. Antibiotics 2023, 12, 151. [Google Scholar] [CrossRef] [PubMed]
- Samedova, A.A.; Kasumov, K.M. Mechanism of action of macrolide antibiotic filipin on cell and lipid membranes. Antibiot. Khimioter. 2009, 54, 44–52. [Google Scholar] [PubMed]
- Ostroumova, O.S.; Efimova, S.S.; Chulkov, E.G.; Schagina, L.V. The interaction of dipole modifiers with polyene-sterol complexes. PLoS ONE 2012, 7, e45135. [Google Scholar] [CrossRef]
- Campagna, S.; Saint, N.; Molle, G.; Aumelas, A. Structure and mechanism of action of the antimicrobial peptide piscidin. Biochemistry 2007, 46, 1771–1778. [Google Scholar] [CrossRef]
- Tevyashova, A.N.; Efimova, S.S.; Alexandrov, A.I.; Ghazy, E.S.M.O.; Bychkova, E.N.; Solovieva, S.E.; Zatonsky, G.B.; Grammatikova, N.E.; Dezhenkova, L.G.; Pereverzeva, E.R.; et al. Semisynthetic Amides of Polyene Antibiotic Natamycin. ACS Infect. Dis. 2023, 9, 42–55. [Google Scholar] [CrossRef]
- Andreoli, T.E. The structure and function of amphotericin B-cholesterol pores in lipid bilayer membranes. Ann. N. Y. Acad. Sci. 1974, 235, 448–468. [Google Scholar] [CrossRef]
- De Kruijff, B.; Gerritsen, W.J.; Oerlemans, A.; Demel, R.A.; van Deenen, L.L. Polyene antibiotic-sterol interactions in membranes of Acholeplasma laidlawii cells and lecithin liposomes. I. Specificity of the membrane permeability changes induced by the polyene antibiotics. Biochim. Biophys. Acta 1974, 339, 30–43. [Google Scholar] [CrossRef]
- Cohen, B.E. Amphotericin B membrane action: Role for two types of ion channels in eliciting cell survival and lethal effects. J. Membr. Biol. 2010, 238, 1–20. [Google Scholar] [CrossRef]
- Dos Santos, A.G.; Marquês, J.T.; Carreira, A.C.; Castro, I.R.; Viana, A.S.; Mingeot-Leclercq, M.P.; de Almeida, R.F.M.; Silva, L.C. The molecular mechanism of Nystatin action is dependent on the membrane biophysical properties and lipid composition. Phys. Chem. Chem. Phys. 2017, 19, 30078–30088. [Google Scholar] [CrossRef] [PubMed]
- Baghirova, A.A.; Kasumov, K.M. Antifungal Macrocycle Antibiotic Amphotericin B-Its Present and Future. Multidisciplinary Perspective for the Use in the Medical Practice. Biochem. Mosc. Suppl. B Biomed. Chem. 2022, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chulkov, E.G.; Schagina, L.V.; Ostroumova, O.S. Membrane dipole modifiers modulate single-length nystatin channels via reducing elastic stress in the vicinity of the lipid mouth of a pore. Biochim. Biophys. Acta 2015, 1848, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Umegawa, Y.; Yamamoto, T.; Dixit, M.; Funahashi, K.; Seo, S.; Nakagawa, Y.; Suzuki, T.; Matsuoka, S.; Tsuchikawa, H.; Hanashima, S.; et al. Amphotericin B assembles into seven-molecule ion channels: An NMR and molecular dynamics study. Sci. Adv. 2022, 8, eabo2658. [Google Scholar] [CrossRef]
- Akkerman, V.; Scheidt, H.A.; Reinholdt, P.; Bashawat, M.; Szomek, M.; Lehmann, M.; Wessig, P.; Covey, D.F.; Kongsted, J.; Müller, P.; et al. Natamycin interferes with ergosterol-dependent lipid phases in model membranes. BBA Adv. 2023, 4, 100102. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Sumino, A.; Shimada, E.; Kinoshita, M.; Matsumori, N.; Oiki, S. Channel Formation and Membrane Deformation via Sterol-Aided Polymorphism of Amphidinol 3. Sci. Rep. 2017, 7, 10782. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.S.; Lee, J.; Lee, D.G. Fungicidal effect of piscidin on Candida albicans: Pore formation in lipid vesicles and activity in fungal membranes. Biol. Pharm. Bull. 2008, 31, 1906–1910. [Google Scholar] [CrossRef]
- Gomes, I.P.; Santos, T.L.; de Souza, A.N.; Nunes, L.O.; Cardoso, G.A.; Matos, C.O.; Costa, L.M.F.; Lião, L.M.; Resende, J.M.; Verly, R.M. Membrane interactions of the anuran antimicrobial peptide HSP1-NH2: Different aspects of the association to anionic and zwitterionic biomimetic systems. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183449. [Google Scholar] [CrossRef]
- Stock, S.D.; Hama, H.; Radding, J.A.; Young, D.A.; Takemoto, J.Y. Syringomycin E inhibition of Saccharomyces cerevisiae: Requirement for biosynthesis of sphingolipids with very-long-chain fatty acids and mannose- and phosphoinositol-containing head groups. Antimicrob. Agents Chemother. 2000, 44, 1174–1180. [Google Scholar] [CrossRef]
- Grilley, M.M.; Stock, S.D.; Dickson, R.C.; Lester, R.L.; Takemoto, J.Y. Syringomycin action gene SYR2 is essential for sphingolipid 4-hydroxylation in Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 11062–11068. [Google Scholar] [CrossRef]
- Bento-Oliveira, A.; Santos, F.C.; Marquês, J.T.; Paulo, P.M.R.; Korte, T.; Herrmann, A.; Marinho, H.S.; de Almeida, R.F.M. Yeast Sphingolipid-Enriched Domains and Membrane Compartments in the Absence of Mannosyldiinositolphosphorylceramide. Biomolecules 2020, 10, 871. [Google Scholar] [CrossRef] [PubMed]
- Idkowiak-Baldys, J.; Grilley, M.M.; Takemoto, J.Y. Sphingolipid C4 hydroxylation influences properties of yeast detergent-insoluble glycolipid-enriched membranes. FEBS Lett. 2004, 569, 272–276. [Google Scholar] [CrossRef]
- Kaulin, Y.A.; Takemoto, J.Y.; Schagina, L.V.; Ostroumova, O.S.; Wangspa, R.; Teeter, J.H.; Brand, J.G. Sphingolipids influence the sensitivity of lipid bilayers to fungicide, syringomycin E. J. Bioenerg. Biomembr. 2005, 37, 339–348. [Google Scholar] [CrossRef]
- Efimova, S.S.; Zakharova, A.A.; Schagina, L.V.; Ostroumova, O.S. Two types of syringomycin E channels in sphingomyelin-containing bilayers. Eur. Biophys. J. 2016, 45, 91–98. [Google Scholar] [CrossRef]
- Mbongo, N.; Loiseau, P.M.; Billion, M.A.; Robert-Gero, M. Mechanism of amphotericin B resistance in Leishmania donovani promastigotes. Antimicrob. Agents Chemother. 1998, 42, 352–357. [Google Scholar] [CrossRef]
- Young, L.Y.; Hull, C.M.; Heitman, J. Disruption of ergosterol biosynthesis confers resistance to amphotericin B in Candida lusitaniae. Antimicrob. Agents Chemother. 2003, 47, 2717–2724. [Google Scholar] [CrossRef]
- Aloia, R.C.; Jensen, F.C.; Curtain, C.C.; Mobley, P.W.; Gordon, L.M. Lipid composition and fluidity of the human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 1988, 85, 900–904. [Google Scholar] [CrossRef]
- Kalvodova, L.; Sampaio, J.L.; Cordo, S.; Ejsing, C.S.; Shevchenko, A.; Simons, K. The lipidomes of vesicular stomatitis virus, semliki forest virus, and the host plasma membrane analyzed by quantitative shotgun mass spectrometry. J. Virol. 2009, 83, 7996–8003. [Google Scholar] [CrossRef]
- Merz, A.; Long, G.; Hiet, M.S.; Brügger, B.; Chlanda, P.; Andre, P.; Wieland, F.; Krijnse-Locker, J.; Bartenschlager, R. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 2011, 286, 3018–3032. [Google Scholar] [CrossRef]
- Gerl, M.J.; Sampaio, J.L.; Urban, S.; Kalvodova, L.; Verbavatz, J.M.; Binnington, B.; Lindemann, D.; Lingwood, C.A.; Shevchenko, A.; Schroeder, C.; et al. Quantitative analysis of the lipidomes of the influenza virus envelope and MDCK cell apical membrane. J. Cell Biol. 2012, 196, 213–221. [Google Scholar] [CrossRef]
- Martín-Acebes, M.A.; Merino-Ramos, T.; Blázquez, A.B.; Casas, J.; Escribano-Romero, E.; Sobrino, F.; Saiz, J.C. The composition of West Nile virus lipid envelope unveils a role of sphingolipid metabolism in flavivirus biogenesis. J. Virol. 2014, 88, 12041–12054. [Google Scholar] [CrossRef]
- Hofmann, S.; Krajewski, M.; Scherer, C.; Scholz, V.; Mordhorst, V.; Truschow, P.; Schöbel, A.; Reimer, R.; Schwudke, D.; Herker, E. Complex lipid metabolic remodeling is required for efficient hepatitis C virus replication. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1041–1056. [Google Scholar] [CrossRef] [PubMed]
- Martín-Acebes, M.A.; Vázquez-Calvo, Á.; Saiz, J.C. Lipids and flaviviruses, present and future perspectives for the control of dengue, Zika, and West Nile viruses. Prog. Lipid Res. 2016, 64, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Mariewskaya, K.A.; Tyurin, A.P.; Chistov, A.A.; Korshun, V.A.; Alferova, V.A.; Ustinov, A.V. Photosensitizing Antivirals. Molecules 2021, 26, 3971. [Google Scholar] [CrossRef]
- Mariewskaya, K.A.; Krasilnikov, M.S.; Korshun, V.A.; Ustinov, A.V.; Alferova, V.A. Near-Infrared Dyes: Towards Broad-Spectrum Antivirals. Int. J. Mol. Sci. 2022, 24, 188. [Google Scholar] [CrossRef] [PubMed]
- Schinazi, R.F.; Chu, C.K.; Babu, J.R.; Oswald, B.J.; Saalmann, V.; Cannon, D.L.; Eriksson, B.F.; Nasr, M. Anthraquinones as a new class of antiviral agents against human immunodeficiency virus. Antivir. Res. 1990, 13, 265–272. [Google Scholar] [CrossRef]
- Tang, J.; Colacino, J.M.; Larsen, S.H.; Spitzer, W. Virucidal activity of hypericin against enveloped and non-enveloped DNA and RNA viruses. Antivir. Res. 1990, 13, 313–325. [Google Scholar] [CrossRef]
- Kraus, G.A.; Pratt, D.; Tossberg, J.; Carpenter, S. Antiretroviral activity of synthetic hypericin and related analogs. Biochem. Biophys. Res. Commun. 1990, 172, 149–153. [Google Scholar] [CrossRef]
- Hudson, J.B.; Imperial, V.; Haugland, R.P.; Diwu, Z. Antiviral activities of photoactive perylenequinones. Photochem. Photobiol. 1997, 65, 352–354. [Google Scholar] [CrossRef]
- Andersen, D.O.; Weber, N.D.; Wood, S.G.; Hughes, B.G.; Murray, B.K.; North, J.A. In vitro virucidal activity of selected anthraquinones and anthraquinone derivatives. Antivir. Res. 1991, 16, 185–196. [Google Scholar] [CrossRef]
- Hudson, J.B.; Lopez-Bazzocchi, I.; Towers, G.H. Antiviral activities of hypericin. Antivir. Res. 1991, 15, 101–112. [Google Scholar] [CrossRef]
- Cohen, P.A.; Hudson, J.B.; Towers, G.H. Antiviral activities of anthraquinones, bianthrones and hypericin derivatives from lichens. Experientia 1996, 52, 180–183. [Google Scholar] [CrossRef]
- Hudson, J.B.; Delaey, E.; de Witte, P.A. Bromohypericins Are Potent Photoactive Antiviral Agents. Photochem. Photobiol. 1999, 70, 820–822. [Google Scholar] [CrossRef]
- Laille, M.; Gerald, F.; Debitus, C. In vitro antiviral activity on dengue virus of marine natural products. Cell. Mol. Life Sci. 1998, 54, 167–170. [Google Scholar] [CrossRef]
- Laurent, D.; Baumann, F.; Benoit, A.G.; Mortelecqe, A.; Nitatpattana, N.; Desvignes, I.; Debitus, C.; Laille, M.; Gonzalez, J.P.; Chungue, E. Structure-activity relationships of dengue antiviral polycyclic quinones. Southeast Asian J. Trop. Med. Public Health 2005, 36, 901–905. [Google Scholar]
- Hudson, J.B.; Zhou, J.; Chen, J.; Harris, L.; Yip, L.; Towers, G.H. Hypocrellin, from Hypocrella bambuase, is phototoxic to human immunodeficiency virus. Photochem. Photobiol. 1994, 60, 253–255. [Google Scholar] [CrossRef]
- Hirayama, J.; Ikebuchi, K.; Abe, H.; Kwon, K.W.; Ohnishi, Y.; Horiuchi, M.; Shinagawa, M.; Ikuta, K.; Kamo, N.; Sekiguchi, S. Photoinactivation of virus infectivity by hypocrellin A. Photochem. Photobiol. 1997, 66, 697–700. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, Y.L.; Xu, C.P.; Gao, J.; Feng, Y.; Wu, Q.F. Disinfection of influenza a viruses by Hypocrellin a-mediated photodynamic inactivation. Photodiagnosis Photodyn. Ther. 2023, 43, 103674. [Google Scholar] [CrossRef]
- St Vincent, M.R.; Colpitts, C.C.; Ustinov, A.V.; Muqadas, M.; Joyce, M.A.; Barsby, N.L.; Epand, R.F.; Epand, R.M.; Khramyshev, S.A.; Valueva, O.A.; et al. Rigid amphipathic fusion inhibitors, small molecule antiviral compounds against enveloped viruses. Proc. Natl. Acad. Sci. USA 2010, 107, 17339–17344. [Google Scholar] [CrossRef]
- Colpitts, C.C.; Ustinov, A.V.; Epand, R.F.; Epand, R.M.; Korshun, V.A.; Schang, L.M. 5-(Perylen-3-yl)ethynyl-arabino-uridine (aUY11), an arabino-based rigid amphipathic fusion inhibitor, targets virion envelope lipids to inhibit fusion of influenza virus, hepatitis C virus, and other enveloped viruses. J. Virol. 2013, 87, 3640–3654. [Google Scholar] [CrossRef]
- Speerstra, S.; Chistov, A.A.; Proskurin, G.V.; Aralov, A.V.; Ulashchik, E.A.; Streshnev, P.P.; Shmanai, V.V.; Korshun, V.A.; Schang, L.M. Antivirals acting on viral envelopes via biophysical mechanisms of action. Antivir. Res. 2018, 149, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Vigant, F.; Hollmann, A.; Lee, J.; Santos, N.C.; Jung, M.E.; Lee, B. The rigid amphipathic fusion inhibitor dUY11 acts through photosensitization of viruses. J. Virol. 2014, 88, 1849–1853. [Google Scholar] [CrossRef]
- Orlov, A.A.; Chistov, A.A.; Kozlovskaya, L.I.; Ustinov, A.V.; Korshun, V.A.; Karganova, G.G.; Osolodkin, D.I. Rigid amphipathic nucleosides suppress reproduction of the tick-borne encephalitis virus. Med. Chem. Commun. 2016, 7, 495–499. [Google Scholar] [CrossRef]
- Chistov, A.A.; Chumakov, S.P.; Mikhnovets, I.E.; Nikitin, T.D.; Slesarchuk, N.A.; Uvarova, V.I.; Rubekina, A.A.; Nikolaeva, Y.V.; Radchenko, E.V.; Khvatov, E.V.; et al. 5-(Perylen-3-ylethynyl)uracil as an antiviral scaffold: Potent suppression of enveloped virus reproduction by 3-methyl derivatives In vitro. Antivir. Res. 2023, 209, 105508. [Google Scholar] [CrossRef]
- Hakobyan, A.; Galindo, I.; Nañez, A.; Arabyan, E.; Karalyan, Z.; Chistov, A.A.; Streshnev, P.P.; Korshun, V.A.; Alonso, C.; Zakaryan, H. Rigid amphipathic fusion inhibitors demonstrate antiviral activity against African swine fever virus. J. Gen. Virol. 2018, 99, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Wiehe, A.; O’Brien, J.M.; Senge, M.O. Trends and targets in antiviral phototherapy. Photochem. Photobiol. Sci. 2019, 18, 2565–2612. [Google Scholar] [CrossRef] [PubMed]
- Straková, P.; Bednář, P.; Kotouček, J.; Holoubek, J.; Fořtová, A.; Svoboda, P.; Štefánik, M.; Huvarová, I.; Šimečková, P.; Mašek, J.; et al. Antiviral activity of singlet oxygen-photogenerating perylene compounds against SARS-CoV-2: Interaction with the viral envelope and photodynamic virion inactivation. Virus Res. 2023, 334, 199158. [Google Scholar] [CrossRef]
- Chistov, A.A.; Orlov, A.A.; Streshnev, P.P.; Slesarchuk, N.A.; Aparin, I.O.; Rathi, B.; Brylev, V.A.; Kutyakov, S.V.; Mikhura, I.V.; Ustinov, A.V.; et al. Compounds based on 5-(perylen-3-ylethynyl)uracil scaffold: High activity against tick-borne encephalitis virus and non-specific activity against enterovirus A. Eur. J. Med. Chem. 2019, 171, 93–103. [Google Scholar] [CrossRef]
- Mariewskaya, K.A.; Gvozdev, D.A.; Chistov, A.A.; Straková, P.; Huvarová, I.; Svoboda, P.; Kotouček, J.; Ivanov, N.M.; Krasilnikov, M.S.; Zhitlov, M.Y.; et al. Membrane-Targeting Perylenylethynylphenols Inactivate Medically Important Coronaviruses via the Singlet Oxygen Photogeneration Mechanism. Molecules 2023, 28, 6278. [Google Scholar] [CrossRef]
- Carpenter, B.L.; Situ, X.; Scholle, F.; Bartelmess, J.; Weare, W.W.; Ghiladi, R.A. Antiviral, Antifungal and Antibacterial Activities of a BODIPY-Based Photosensitizer. Molecules 2015, 20, 10604–10621. [Google Scholar] [CrossRef]
- Wolf, M.C.; Freiberg, A.N.; Zhang, T.; Akyol-Ataman, Z.; Grock, A.; Hong, P.W.; Li, J.; Watson, N.F.; Fang, A.Q.; Aguilar, H.C.; et al. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl. Acad. Sci. USA 2010, 107, 3157–3162. [Google Scholar] [CrossRef]
- Vigant, F.; Lee, J.; Hollmann, A.; Tanner, L.B.; Akyol Ataman, Z.; Yun, T.; Shui, G.; Aguilar, H.C.; Zhang, D.; Meriwether, D.; et al. A mechanistic paradigm for broad-spectrum antivirals that target virus-cell fusion. PLoS Pathog. 2013, 9, e1003297. [Google Scholar] [CrossRef]
- Hollmann, A.; Gonçalves, S.; Augusto, M.T.; Castanho, M.A.; Lee, B.; Santos, N.C. Effects of singlet oxygen generated by a broad-spectrum viral fusion inhibitor on membrane nanoarchitecture. Nanomedicine 2015, 11, 1163–1167. [Google Scholar] [CrossRef]
- Arnaut, Z.A.; Pinto, S.M.A.; Aroso, R.T.; Amorim, A.S.; Lobo, C.S.; Schaberle, F.A.; Pereira, D.; Núñez, J.; Nunes, S.C.C.; Pais, A.A.C.C.; et al. Selective, broad-spectrum antiviral photodynamic disinfection with dicationic imidazolyl chlorin photosensitizers. Photochem. Photobiol. Sci. 2023, 22, 2607–2620. [Google Scholar] [CrossRef] [PubMed]
- Jurak, I.; Cokarić Brdovčak, M.; Djaković, L.; Bertović, I.; Knežević, K.; Lončarić, M.; Jurak Begonja, A.; Malatesti, N. Photodynamic Inhibition of Herpes Simplex Virus 1 Infection by Tricationic Amphiphilic Porphyrin with a Long Alkyl Chain. Pharmaceutics 2023, 15, 956. [Google Scholar] [CrossRef]
- Monjo, A.L.; Pringle, E.S.; Thornbury, M.; Duguay, B.A.; Monro, S.M.A.; Hetu, M.; Knight, D.; Cameron, C.G.; McFarland, S.A.; McCormick, C. Photodynamic Inactivation of Herpes Simplex Viruses. Viruses 2018, 10, 532. [Google Scholar] [CrossRef] [PubMed]
- Meunier, T.; Desmarets, L.; Bordage, S.; Bamba, M.; Hervouet, K.; Rouillé, Y.; François, N.; Decossas, M.; Sencio, V.; Trottein, F.; et al. A Photoactivable Natural Product with Broad Antiviral Activity against Enveloped Viruses, Including Highly Pathogenic Coronaviruses. Antimicrob. Agents Chemother. 2022, 66, e0158121. [Google Scholar] [CrossRef] [PubMed]
- Nikolayeva, Y.V.; Ulashchik, E.A.; Chekerda, E.V.; Galochkina, A.V.; Slesarchuk, N.A.; Chistov, A.A.; Nikitin, T.D.; Korshun, V.A.; Shmanai, V.V.; Ustinov, A.V.; et al. 5-(Perylen-3-ylethynyl)uracil Derivatives Inhibit Reproduction of Respiratory Viruses. Russ. J. Bioorg. Chem. 2020, 46, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Proskurin, G.V.; Orlov, A.A.; Brylev, V.A.; Kozlovskaya, L.I.; Chistov, A.A.; Karganova, G.G.; Palyulin, V.A.; Osolodkin, D.I.; Korshun, V.A.; Aralov, A.V. 3′-O-Substituted 5-(perylen-3-ylethynyl)-2′-deoxyuridines as tick-borne encephalitis virus reproduction inhibitors. Eur. J. Med. Chem. 2018, 155, 77–83. [Google Scholar] [CrossRef]
- Weil, T.; Groß, R.; Röcker, A.; Bravo-Rodriguez, K.; Heid, C.; Sowislok, A.; Le, M.H.; Erwin, N.; Dwivedi, M.; Bart, S.M.; et al. Supramolecular Mechanism of Viral Envelope Disruption by Molecular Tweezers. J. Am. Chem. Soc. 2020, 142, 17024–17038. [Google Scholar] [CrossRef]
- Lump, E.; Castellano, L.M.; Meier, C.; Seeliger, J.; Erwin, N.; Sperlich, B.; Stürzel, C.M.; Usmani, S.; Hammond, R.M.; von Einem, J.; et al. A molecular tweezer antagonizes seminal amyloids and HIV infection. Elife 2015, 4, e05397. [Google Scholar] [CrossRef]
- Röcker, A.E.; Müller, J.A.; Dietzel, E.; Harms, M.; Krüger, F.; Heid, C.; Sowislok, A.; Riber, C.F.; Kupke, A.; Lippold, S.; et al. The molecular tweezer CLR01 inhibits Ebola and Zika virus infection. Antivir. Res. 2018, 152, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Weil, T.; Kirupakaran, A.; Le, M.H.; Rebmann, P.; Mieres-Perez, J.; Issmail, L.; Conzelmann, C.; Müller, J.A.; Rauch, L.; Gilg, A.; et al. Advanced Molecular Tweezers with Lipid Anchors against SARS-CoV-2 and Other Respiratory Viruses. JACS Au 2022, 2, 2187–2202. [Google Scholar] [CrossRef]
- Wang, G.; Watson, K.M.; Buckheit, R.W., Jr. Anti-human immunodeficiency virus type 1 activities of antimicrobial peptides derived from human and bovine cathelicidins. Antimicrob. Agents Chemother. 2008, 52, 3438–3440. [Google Scholar] [CrossRef]
- Kagan, B.L.; Ganz, T.; Lehrer, R.I. Defensins: A family of antimicrobial and cytotoxic peptides. Toxicology 1994, 87, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liu, Z.; Peng, H.; Lv, Y.; Feng, Y.; Kang, J.; Lu, N.; Ma, R.; Hou, S.; Sun, W.; et al. Bomidin: An Optimized Antimicrobial Peptide with Broad Antiviral Activity Against Enveloped Viruses. Front. Immunol. 2022, 13, 851642. [Google Scholar] [CrossRef] [PubMed]
- Omer, A.A.M.; Hinkula, J.; Tran, P.T.; Melik, W.; Zattarin, E.; Aili, D.; Selegård, R.; Bengtsson, T.; Khalaf, H. Plantaricin NC8 αβ rapidly and efficiently inhibits flaviviruses and SARS-CoV-2 by disrupting their envelopes. PLoS ONE 2022, 17, e0278419. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.T.; Huang, Y.H.; Rosengren, K.J.; Franquelim, H.G.; Carvalho, F.A.; Johnson, A.; Sonza, S.; Tachedjian, G.; Castanho, M.A.; Daly, N.L.; et al. Decoding the membrane activity of the cyclotide kalata B1: The importance of phosphatidylethanolamine phospholipids and lipid organization on hemolytic and anti-HIV activities. J. Biol. Chem. 2011, 286, 24231–24241. [Google Scholar] [CrossRef]
- Kozlov, M.M.; Leikin, S.L.; Chernomordik, L.V.; Markin, V.S.; Chizmadzhev, Y.A. Stalk mechanism of vesicle fusion. Intermixing of aqueous contents. Eur. Biophys. J. 1989, 17, 121–129. [Google Scholar] [CrossRef]
- Cooke, I.R.; Deserno, M. Coupling between lipid shape and membrane curvature. Biophys. J. 2006, 91, 487–495. [Google Scholar] [CrossRef]
- Miao, L.; Stafford, A.; Nir, S.; Turco, S.J.; Flanagan, T.D.; Epand, R.M. Potent inhibition of viral fusion by the lipophosphoglycan of Leishmania donovani. Biochemistry 1995, 34, 4676–4683. [Google Scholar] [CrossRef] [PubMed]
- Rasmusson, B.J.; Flanagan, T.D.; Turco, S.J.; Epand, R.M.; Petersen, N.O. Fusion of Sendai virus and individual host cells and inhibition of fusion by lipophosphoglycan measured with image correlation spectroscopy. Biochim. Biophys. Acta 1998, 1404, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Sardar, A.; Lahiri, A.; Kamble, M.; Mallick, A.I.; Tarafdar, P.K. Translation of Mycobacterium Survival Strategy to Develop a Lipo-peptide based Fusion Inhibitor. Angew. Chem. Int. Ed. Engl. 2021, 60, 6101–6106. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Zhang, S.; Wang, Y.; Li, Y.; Wang, X.; Yang, Q. Surfactin Inhibits Membrane Fusion during Invasion of Epithelial Cells by Enveloped Viruses. J. Virol. 2018, 92, e00809-18. [Google Scholar] [CrossRef]
- Kracht, M.; Rokos, H.; Ozel, M.; Kowall, M.; Pauli, G.; Vater, J. Antiviral and hemolytic activities of surfactin isoforms and their methyl ester derivatives. J. Antibiot. 1999, 52, 613–619. [Google Scholar] [CrossRef]
- Shekunov, E.V.; Zlodeeva, P.D.; Efimova, S.S.; Muryleva, A.A.; Zarubaev, V.V.; Slita, A.V.; Ostroumova, O.S. Cyclic lipopeptides as membrane fusion inhibitors against SARS-CoV-2: New tricks for old dogs. Antivir. Res. 2023, 212, 105575. [Google Scholar] [CrossRef]
- Shekunov, E.V.; Efimova, S.S.; Yudintceva, N.M.; Muryleva, A.A.; Zarubaev, V.V.; Slita, A.V.; Ostroumova, O.S. Plant Alkaloids Inhibit Membrane Fusion Mediated by Calcium and Fragments of MERS-CoV and SARS-CoV/SARS-CoV-2 Fusion Peptides. Biomedicines 2021, 9, 1434. [Google Scholar] [CrossRef]








| Inhibitor | Structure | Enzyme | Origin | IC50, μM | References |
|---|---|---|---|---|---|
| amino-oxazole dibenzylamide |  | AccC | E. coli | 0.125 | [3] |
| (R)-2-(2-chlorobenzylamino)-1-(2,3-dihydro-1H-inden-1-yl)-1H-imidazo[4,5-b]pyridine-5-carboxamide |  | AccC | E. coli | 0.02 | [4] |
| moiramide B |  | AccAD | S. aureus | 0.096 | [5] |
| E. coli | 0.006 | [5] | |||
| andrimid |  | AccAD | S. aureus | 0.091 | [5] |
| E. coli | 0.004 | [5] | |||
| thiolactomycin |  | FabH | S. pneumoniae | 7.9 ± 1.1 | [6] |
| H. influenzae | 5.8 ± 1.6 | [6] | |||
| M. tuberculosis | 24 | [7] | |||
| E. coli | 32–110 | [6,8] | |||
| FabF | E. coli | 6 | [8] | ||
| FabB | E. coli | 2–25 | [8,9] | ||
| SB418011 |  | FabH | S. pneumoniae | 0.016 ± 0.003 | [6] |
| H. influenzae | 0.59 ± 0.05 | [6] | |||
| E. coli | 1.20 ± 0.40 | [6] | |||
| cerulenin |  | FabF | E. coli | 20 | [8] |
| FabB | E. coli | 3 | [8] | ||
| platensimycin |  | FabF | S. aureus | 0.02–0.29 | [10,11] |
| E. coli | 0.02 | [12] | |||
| platencin |  | FabH | S. aureus | 9.2–16.2 | [10,11] |
| FabF | S. aureus | 0.1–4.6 | [10,11] | ||
| (-)-epigallocatechin gallate |  | FabG | E. coli | 5 | [13] |
| P. falciparum | 0.3 | [14,15] | |||
| FabI | E. coli | 15 | [13] | ||
| P. falciparum | 0.2 | [14] | |||
| FabZ | P. falciparum | 0.03–0.4 | [14,15] | ||
| (-)-gallocatechin gallate |  | FabG | E. coli | 10 | [13] |
| P. falciparum | 1.1 | [14] | |||
| FabI | E. coli | 5 | [13] | ||
| P. falciparum | 0.5 | [14] | |||
| FabZ | P. falciparum | 0.6 | [14] | ||
| (-)-epicatechin gallate |  | FabG | E. coli | 15 | [13] |
| P. falciparum | 1 | [14] | |||
| FabI | E. coli | 10 | [13] | ||
| P. falciparum | 0.2 | [14] | |||
| FabZ | P. falciparum | 0.4 | [14] | ||
| (-)-catechin gallate |  | FabG | E. coli | 10 | [13] |
| P. falciparum | 1 | [14] | |||
| FabI | E. coli | 5 | [13] | ||
| P. falciparum | 0.3 | [14] | |||
| FabZ | P. falciparum | 0.4 | [14] | ||
| butein |  | FabG | E. coli | 10 | [13] |
| FabI | E. coli | 30 | [13] | ||
| isoliquiritigenin |  | FabG | E. coli | 20 | [13] |
| FabI | E. coli | 40 | [13] | ||
| 2,2′,4′-trihydroxychalcone |  | FabG | E. coli | 25 | [13] |
| FabI | E. coli | 40 | [13] | ||
| fisetin |  | FabG | E. coli | 30 | [13] |
| P. falciparum | 4.1 | [14] | |||
| FabI | E. coli | 50 | [13] | ||
| P. falciparum | 1 | [14] | |||
| FabZ | P. falciparum | 2 | [14] | ||
| quercetin |  | FabG | E. coli | 20 | [13] |
| P. falciparum | 5.4 | [14] | |||
| FabI | E. coli | 20 | [13] | ||
| P. falciparum | 1.5 | [14] | |||
| FabZ | P. falciparum | 1.5 | [14] | ||
| resveratrol |  | FabG | E. coli | 65 | [13] |
| FabI | E. coli | 30 | [13] | ||
| piceatannol |  | FabG | E. coli | 35 | [13] |
| FabI | E. coli | 15 | [13] | ||
| fustin |  | FabG | E. coli | 25 | [13] |
| FabI | E. coli | 40 | [13] | ||
| taxifolin |  | FabG | E. coli | 20 | [13] |
| FabI | E. coli | 30 | [13] | ||
| kaempferol |  | FabG | P. falciparum | 4 | [14] |
| FabI | P. falciparum | 20 | [14] | ||
| luteolin |  | FabG | P. falciparum | 4 | [14] |
| FabI | P. falciparum | 2 | [14] | ||
| FabZ | P. falciparum | 5 | [14] | ||
| luteolin 7-O-β-D-glucopyranoside |  | FabI | P. falciparum | 22 | [16] |
| myricetin |  | FabG | P. falciparum | 14 | [14] |
| FabI | P. falciparum | 0.4 | [14] | ||
| FabZ | P. falciparum | 2 | [14] | ||
| isorhamnetin |  | FabG | P. falciparum | 8.3 | [14] |
| FabI | P. falciparum | 5 | [14] | ||
| 7,3′,4′-trihydroxyisoflavone |  | FabG | E. coli | 35 | [13] |
| FabI | E. coli | 25 | [13] | ||
| morin |  | FabG | P. falciparum | 2.3 | [14] |
| FabI | P. falciparum | 5 | [14] | ||
| FabZ | P. falciparum | 8 | [14] | ||
| macrolactin S |  | FabG | S. aureus | 130 | [17] |
| macrolactin B |  | FabG | S. aureus | 100 | [17] |
| NAS-21 |  | FabZ | M. smegmatis | 360 | [18] |
| NAS-91 |  | FabZ | M. smegmatis | 498 | [18] |
| emodin |  | FabZ | F. tularensis | 43.1 ± 9.2 | [19] |
| Y. pestis | 29.7 ± 6.0 | [19] | |||
| H. pylori | 9.70 ± 1.0 | [20] | |||
| mangostin |  | FabZ | F. tularensis | 7.7 ± 2.0 | [19] |
| Y. pestis | 6.1 ± 1.4 | [19] | |||
| stictic acid |  | FabZ | F. tularensis | 27.8 ± 6.1 | [19] |
| Y. pestis | 13.0 ± 1.4 | [19] | |||
| 1,4-naphthoquinone |  | FabD | M. catarrhalis | 23.18 ± 2.48 | [21] |
| FabZ | M. catarrhalis | 26.67 ± 3.34 | [21] | ||
| juglone |  | FabD | H. pylori | 20 ± 1 | [22] |
| FabZ | F. tularensis | 5.4 ± 1.4 | [19] | ||
| Y. pestis | 5.3 ± 1.0 | [19] | |||
| H. pylori | 30 ± 4 | [22] | |||
| triclosan |  | FabI | E. coli | 0.98 | [23] |
| S. aureus | 0.44–0.66 | [23,24,25] | |||
| P. falciparum | 0.05–2 | [15,26] | |||
| C. trachomatis | 0.32 ± 0.08 | [27] | |||
| AFN-1252 |  | FabI | C. trachomatis | 0.95 ± 0.21 | [27] |
| xanthorrhizol |  | FabI | E. coli | 17.1 ± 1.8 | [28] |
| complestatin |  | FabI | S. aureus | 0.5 | [25] |
| FabK | S. pneumoniae | 10 | [25] | ||
| neuroprotectin A |  | FabI | S. aureus | 0.3 | [25] |
| chloropeptin I |  | FabI | S. aureus | 0.6 | [25] |
| meleagrin |  | FabI | S. aureus | 40.1 | [23] |
| E. coli | 33.2 | [23] | |||
| phellinstatin |  | FabI | S. aureus | 6 | [29] |
| chalcomoracin |  | FabI | S. aureus | 5.5 | [30] |
| moracin C |  | FabI | S. aureus | 83.8 | [30] |
| aquastatin A |  | FabI | S. aureus | 3.2 | [31] |
| FabK | S. pneumoniae | 9.2 | [31] | ||
| atromentin |  | FabK | S. pneumoniae | 0.24 | [32] |
| leucomelone |  | FabK | S. pneumoniae | 1.57 | [32] |
| (Z)-1-oxooctadec-11-enylphosphoramidic acid |  | PlsY | S. pneumoniae | 11 | [33] |
| 1,1-difluoro-2-oxotridecylphosphonic acid |  | PlsY | B. anthracis | 25 | [33] |
| phenyl (8-phenyloctanoyl) sulfamate |  | PlsY | S. aureus | 25 | [34] |
| dioctylamine |  | CfaS | H. pylori | 63.81 | [35] |
| Inhibitor | Structure | Enzyme | Origin | IC50, μM | References |
|---|---|---|---|---|---|
| peptide 920 | NH2-SSGWMLDPIAGKWSR-COOH | LpxA | E. coli | 0.06 ± 0.01 | [206] |
| RJPXD33 | TNLYMLPKWDIP-NH2 | LpxA | E. coli | 19.0 ± 1.2 | [207] |
| LpxD | E. coli | 3.5 ± 0.1 | [207] | ||
| (R)-(3-(2-chloro-6-methoxybenzyl)morpholino)(3-(4-methylpyridin-2-yl)-1H-pyrazol-5-yl)methanone |  | LpxA | E. coli | 0.6 | [208] |
| BB-78485 |  | LpxC | E. coli | 0.16 ± 0.07 | [209] |
| L-161,240 |  | LpxC | E. coli | 0.023 ± 0.003 | [210] |
| P. aeruginosa | 0.22 ± 0.003 | [210] | |||
| L-573,655 |  | LpxC | E. coli | 8.5 | [211] |
| CHIR-090 |  | LpxC | A. aeolicus | ~0.003 | [212] |
| E. coli | 0.009 | [213] | |||
| R. leguminosarum | 0.69 | [213] | |||
| LpxC-4 |  | LpxC | P. aeruginosa | 0.001 | [214] |
| K. pneumoniae | 0.00007 | [214] | |||
| A. baumannii | 0.183 | [214] | |||
| TU-514 |  | LpxC | A. aeolicus | 7.0 ± 0.5 | [215] |
| E. coli | 7.2 ± 1.9 | [215] | |||
| 4-(2-Chlorophenyl)-3-hydroxy-7,7-dimethyl-2-phenyl-6,7,8,9-tetrahydro-2H-pyrazolo[3,4-b]quinolin-5(4H)-one |  | LpxD | E. coli | 3.2 | [216] |
| 1-(5-((4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)sulfonyl)indolin-1-yl)ethan-1-one |  | LpxH | E. coli | 1.2 ± 0.2 | [217] |
| AZ1 |  | LpxH | K. pneumoniae | 0.36 | [218] |
| E. coli | 0.14 | [218,219] | |||
| JH-LPH-28 |  | LpxH | K. pneumoniae | 0.11 | [218] |
| E. coli | 0.083 | [218] | |||
| JH-LPH-33 |  | LpxH | K. pneumoniae | 0.026 | [218] |
| E. coli | 0.046 | [218] |
| Agent | Structure | Cmin, μM | Ctr, μM | Lipid Composition | References |
|---|---|---|---|---|---|
| Pore formation | |||||
| gramicidin A |  | 0.001 | – * | DSPC | [254] |
| alamethicin |  | 0.1 | – * | DOPS:DOPE 1:1 (m/m) | [293] |
| pardaxin |  | 0.006 | – * | soybean lecithin | [258] |
| melittin |  | 0.23 | – * | POPC:cholesterol 3:1 (m/m) | [294] |
| magainin I |  | 10 | – * | DOPC; DOPE:DOPG 3:1 (m/m) | [260] |
| magainin II |  | 0.08 | – * | POPC:DOPG 6:1 (m/m); DOPS:ergosterol 3:1 (m/m); POPC:ergosterol 3:1 (m/m) | [261] |
| mastoparan |  | 0.68 | – * | DPhPC | [262] |
| ceratotoxin A |  | 0.02 | – * | POPC:DOPE 7:3 (w/w); POPC:DOPE:POPS 7:3:1 (w/w) | [271,272] |
| protegrin-1 |  | 0.25–10 | – * | DOPC:DOPE 1:1 (m/m) | [259] |
| nisin |  | 0.1 | lipid II | [295] | |
| ~40 | >500 | TOCL | [265] | ||
| cinnamycin |  | ~1.5 | >10 | DOPE; TOCL | [265] |
| duramycin |  | 4.5–10 | – * | GMO | [266] |
| ~2 | >12 | DOPE; TOCL | [265] | ||
| rabbit α-defensins (NP-1/2) |  | ~1 | >16 | PE/PC/PS 2:2:1 (w/w); PE/CL | [296,297] |
| daptomycin |  | 6.2 | – * | DPhPG | [298] |
| polymyxin B |  | 2.5 | >100 | DOPG | [108] |
| 1 | >20 | Kdo2-Lipid A | |||
| gaysemycin |  | ~26 | – * | DOPG | [279] |
| Pore formation and detergent action | |||||
| cecropin A |  | 1 | >5 | DOPS:DOPE 1:1 (m/m) | [264] |
| cecropin B |  | 1 | >5 | DOPS:DOPE 1:1 (m/m) | [264] |
| Detergent action | |||||
| cecropin P1 |  | – * | >50 | DOPS:DOPE 1:1 (m/m) | [264] |
| aurein 1.2 |  | – * | >10 | DMPG | [287] |
| Inhibitor | Structure | Enzyme | IC50, μM | References | |
|---|---|---|---|---|---|
| sphingofungin B |  | SPT | C. albicans | 0.049 | [350] |
| S. cerevisiae | 0.051 | ||||
| viridiofungin A |  | SPT | C. albicans | 0.022 | [350] |
| S. cerevisiae | 4.7 | ||||
| viridiofungin B |  | SPT | C. albicans | 0.017 | [350] |
| S. cerevisiae | 1.84 | ||||
| viridiofungin C |  | SPT | C. albicans | 0.025 | [350] |
| S. cerevisiae | 1.68 | ||||
| aureobasidin A |  | IPCS | C. albicans | 0.002 | [363] |
| C. glabrata | 0.002 | ||||
| Candida tropicalis | 0.003 | ||||
| Candida parapsilosis | 0.003 | ||||
| Candida krusei | 0.003 | ||||
| A. fumigatus | 0.005 | ||||
| Aspergillus flavus | 0.002 | ||||
| Aspergillus terreus | 0.004 | ||||
| Aspergillus niger | 0.004 | ||||
| S. cerevisiae | 0.0009 | [358] | |||
| khafrefungin |  | IPCS | C. albicans | 0.0006 | [356] |
| C. neoformans | 0.031 | ||||
| S. cerevisiae | 0.007 | ||||
| haplofungin A |  | IPCS | S. cerevisiae | 0.0025 | [357] |
| A. fumigatus | 0.41 | ||||
| haplofungin B |  | IPCS | S. cerevisiae | 0.042 | [357] |
| A. fumigatus | 1.33 | ||||
| pleofungin A |  | IPCS | S. cerevisiae | 0.007 | [358] |
| A. fumigatus | 0.0009 | ||||
| galbonolide A (rustmicin) |  | IPCS | C. albicans | 0.0038 | [359] |
| C. neoformans | 0.00007 | ||||
| S. cerevisiae | 0.0198 |
| Agent | Structure | Cmin, μM | Ctr, μM | Target Lipid | References |
|---|---|---|---|---|---|
| Pore formation | |||||
| syringomycin E |  | 1–5 | – * | DPhPC; DOPS:DOPE 1:1 (m/m) | [422] |
| syringopeptin 22A |  | 0.003 | – * | DPhPC; DOPS:DOPE 1:1 (m/m) | [416] |
| syringopeptin 25A |  | 0.004 | – * | PC:PE:PS 2:2:1 (m/m/m) | [428] |
| fengycins |  | 0.1–0.5 | >10 | POPC:POPE:POPG:ergosterol 2:2:5:1 (m/m) | [425] |
| surfactin |  | 0.2–0.4 | – * | DPhPC | [424] |
| 1.4 | – * | glyceryl monooleate | [426] | ||
| iturin A |  | 0.001 | – * | egg-PC; egg-PC:DMPE 8:2 (v/v) | [429] |
| – * | – * | glyceromonoolein | [421] | ||
| mycosubtilin |  | – * | –* | DPhPC | [430] |
| bacillomycins |  | – * | – * | glyceromonoolein | [421] |
| amphotericin B |  | 0.02–0.03 | – * | phospholipid:cholesterol 20:1 (m/m) | [431] |
| 0.01 | >20 | DPhPC:ergosterol 2:1 (m/m) | [432] | ||
| nystatin |  | 0.1 | – * | phospholipid:cholesterol 20:1 (m/m) | [431] |
| 0.01 | >100 | DPhPC:ergosterol 2:1 (m/m) | [433] | ||
| filipin |  | 0.02 | – * | phospholipid:cholesterol 2:1 (v/v) | [434] |
| 0.01 | >100 | DPhPC:ergosterol 2:1 (m/m) | [435] | ||
| piscidin |  | 0.005 | – * | azolectin | [436] |
| Detergent action | |||||
| natamycin |  | – * | 110 | DPhPC:ergosterol 2:1 (m/m) | [437] |
| Molecular Tweezer | Structure | Virus | IC50, µM | Reference |
|---|---|---|---|---|
| CLR01 |  | HIV-1 | 13.7–20.1 | [501] |
| Ebola | 25.8 | [502] | ||
| Zika | 8.2 | [502] | ||
| HSV-1 | 19.3 | [500] | ||
| HSV-2 | ||||
| measles | ||||
| IVA | ||||
| SARS-CoV-2 | 76.7 | [503] | ||
| CLR05 |  | Zika | 38.1 | [500] |
| HSV-1 | ||||
| HSV-2 | ||||
| measles | ||||
| influenza | ||||
| SARS-CoV-2 | 167.3 | [503] | ||
| CLR01e |  | HIV-1 | ~10 | [500] |
| CLR01f |  | HIV-1 | ~7 | [500] |
| CP006 |  | SARS-CoV-2 | 0.3 | [503] |
| CP020 |  | SARS-CoV-2 | 0.4 | [503] |
| CP025 |  | SARS-CoV-2 | 0.6 | [503] |
| CP036 |  | SARS-CoV-2 | 0.2 | [503] |






{kind=link}
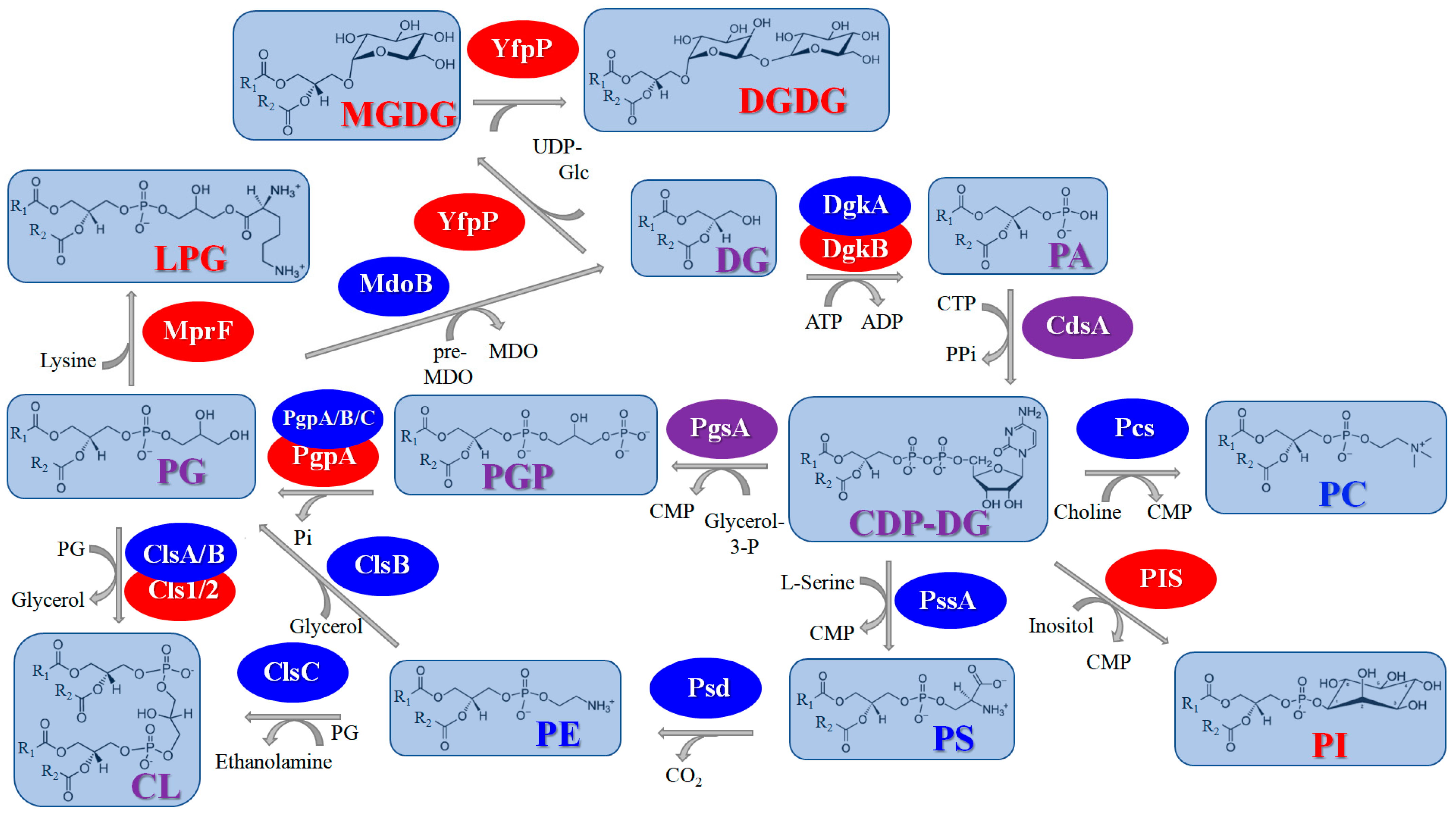{kind=link}
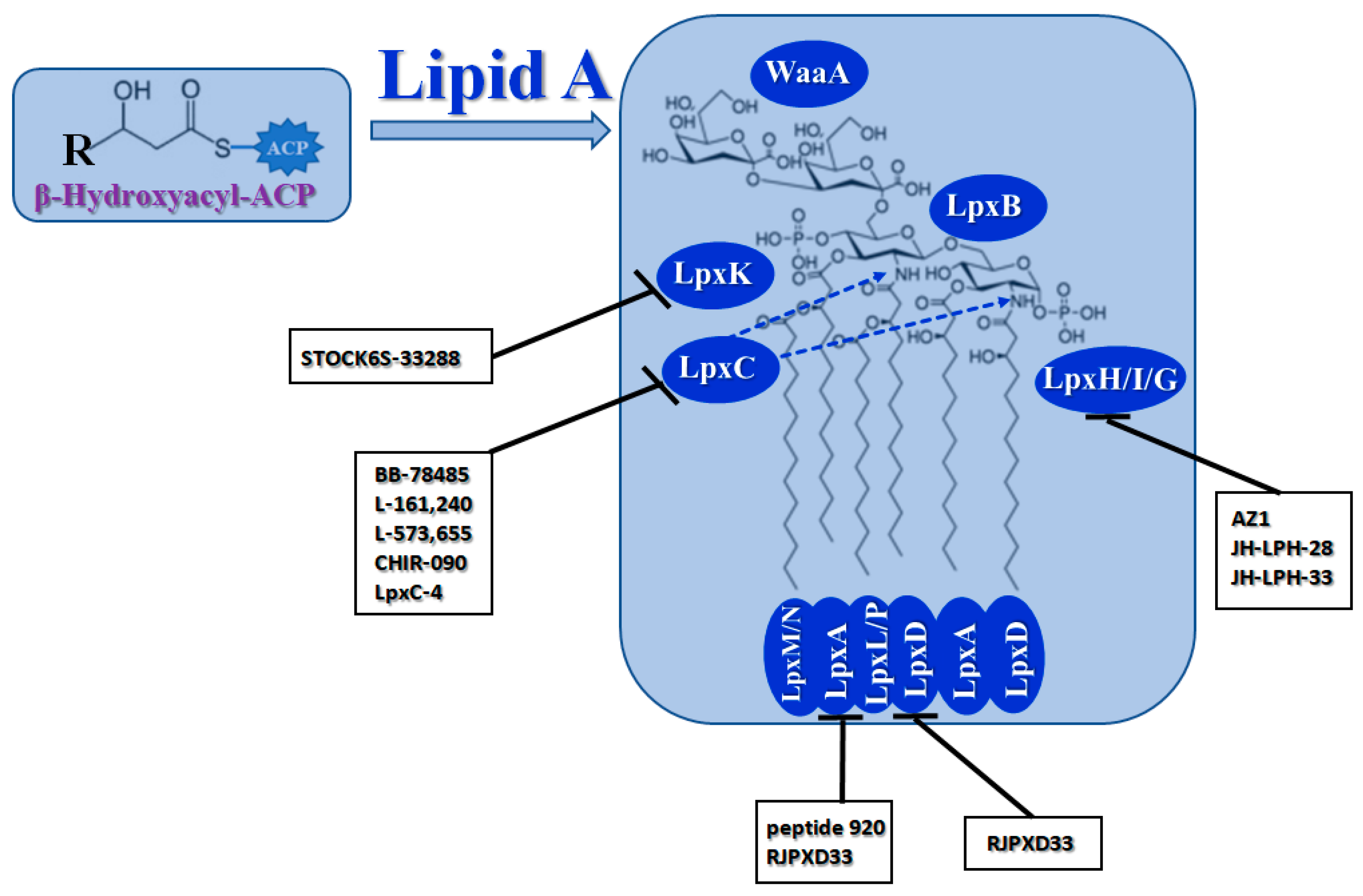{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
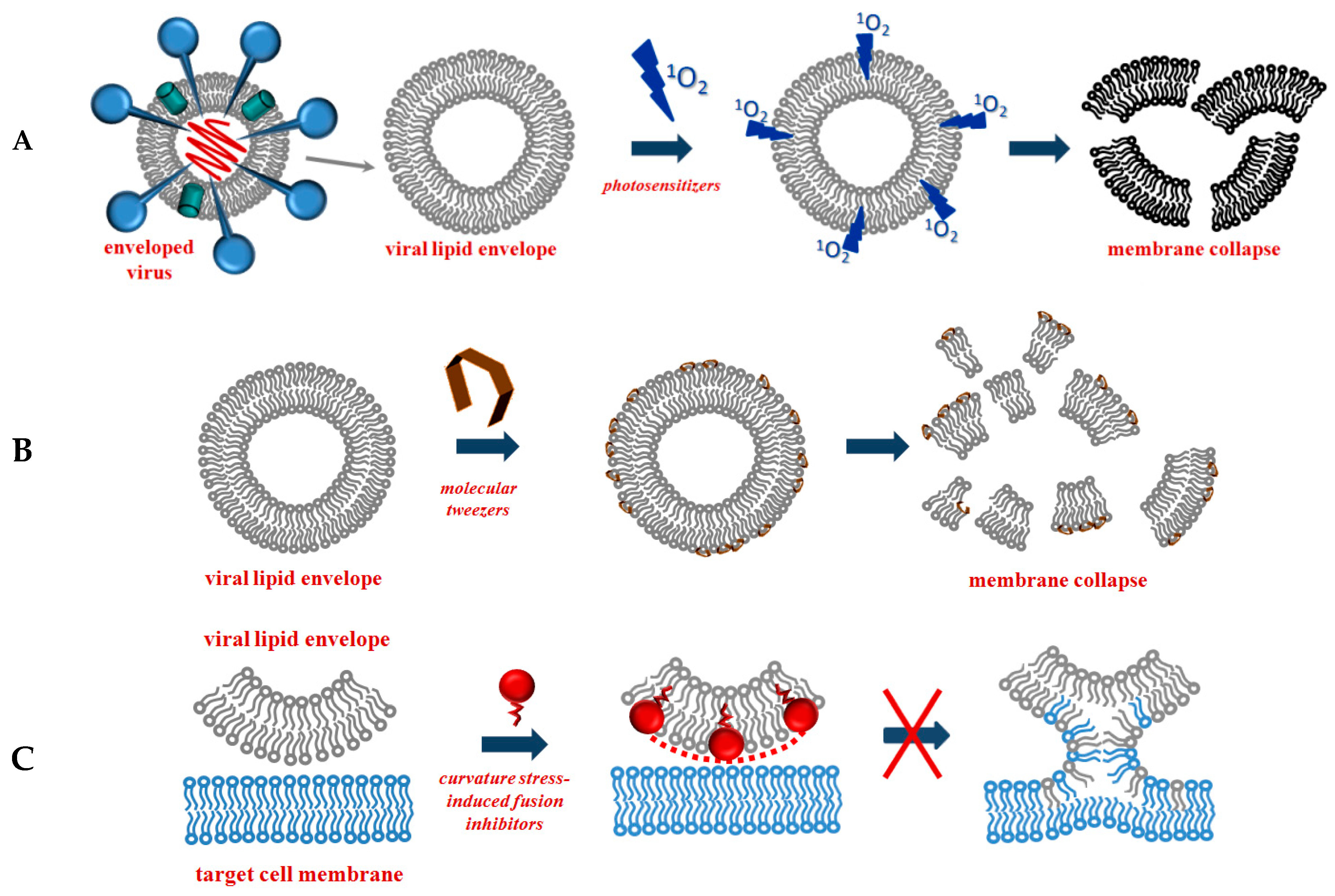{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ostroumova, O.S.; Efimova, S.S. Lipid-Centric Approaches in Combating Infectious Diseases: Antibacterials, Antifungals and Antivirals with Lipid-Associated Mechanisms of Action. Antibiotics 2023, 12, 1716. https://doi.org/10.3390/antibiotics12121716
Ostroumova OS, Efimova SS. Lipid-Centric Approaches in Combating Infectious Diseases: Antibacterials, Antifungals and Antivirals with Lipid-Associated Mechanisms of Action. Antibiotics. 2023; 12(12):1716. https://doi.org/10.3390/antibiotics12121716
Chicago/Turabian StyleOstroumova, Olga S., and Svetlana S. Efimova. 2023. "Lipid-Centric Approaches in Combating Infectious Diseases: Antibacterials, Antifungals and Antivirals with Lipid-Associated Mechanisms of Action" Antibiotics 12, no. 12: 1716. https://doi.org/10.3390/antibiotics12121716