
Abstract
The multifaceted organization of the immune system involves not only patrolling lymphocytes that constantly monitor antigen-presenting cells in secondary lymphoid organs but also immune cells that establish permanent tissue-residency. The integration in the respective tissue and the adaption to the organ milieu enable tissue-resident cells to establish signaling circuits with parenchymal cells to coordinate immune responses and maintain tissue homeostasis. Innate lymphoid cells (ILCs) are tissue-resident innate immune cells that have a similar functional diversity to T cells including lineage-specifying transcription factors that drive certain effector programs. Since their formal discovery 10 years ago, it has become clear that ILCs are present in almost every tissue but strongly enriched at barrier surfaces, where they regulate immunity to infection, chronic inflammation, and tissue maintenance. In this context, recent research has identified ILCs as key in orchestrating tissue homeostasis through their ability to sustain bidirectional interactions with epithelial cells, neurons, stromal cells, adipocytes, and many other tissue-resident cells. In this review, we provide a comprehensive discussion of recent studies that define the development and heterogeneity of ILC populations and their impact on innate and adaptive immunity. Further, we discuss emerging research on the influence of the nervous system, circadian rhythm, and developmental plasticity on ILC function. Uncovering the signaling circuits that control development and function of ILCs will provide an integrated view on how immune responses in tissues are synchronized with functional relevance far beyond the classical view of the role of the immune system in discrimination between self/non-self and host defense.
Similar content being viewed by others
Introduction
The transition of the innate to the adaptive immune response takes place in the secondary lymphoid organs where antigen-presenting cells present peptides via major histocompatibility complex (MHC) molecules to patrolling naïve T cells, which scan numerous cells searching for their specific antigen. While activation of the adaptive immune response in the lymphoid structure is key in fighting infections and forming immunological memory, it also became apparent that diverse immune cells including innate and adaptive lymphocytes establish permanent tissue-residency in peripheral organs thus creating a first-line defense system against invading pathogens at barrier surfaces.1 Among tissue-resident cells, innate lymphoid cells (ILCs) emerge as populations of innate immune cells that have lymphoid morphology but lack rearranged antigen receptors.2 As tissue-resident cells, ILCs adapt to the organ milieu and establish close interactions with hematopoietic cells but also with non-hematopoietic cells in the tissues including neurons, epithelial cells, stromal cells, and other parenchymal cells, such as adipocytes and hepatocytes.3,4 The challenge in the upcoming years will be to uncover the cellular and molecular basis of these tissue-resident circuits, which regulate inflammation and tissue homeostasis in health and disease.
ILCs are classified into different groups depending on their developmental and effector program defined by the expression of lineage-specifying transcription factors (TFs).2,5 The expression of a lineage-specifying TF is selective to a certain degree, sometimes not exclusive though, as in the case of GATA binding protein 3 (GATA-3) or T-box expressed in T cells (T-bet), which are expressed in several ILC lineages. Further, the lineage-specifying factor is essential for the development of the corresponding ILC lineage. Similar to T cells and based on developmental, functional, and migratory aspects, mature ILCs can be divided into cytotoxic (interleukin-7 receptor α−, IL-7Rα–) and non-cytotoxic (or helper-like, IL-7Rα+) ILCs.2,5 Natural killer (NK) cells are the oldest member of the ILC family and the only representative of cytotoxic ILCs. NK cells express the lineage-specifying T-box TF Eomesodermin (Eomes), and mediate effector functions via perforin-dependent cytotoxicity and production of IFN-γ, which together protect from intracellular pathogens and tumors.6,7 Non-cytotoxic ILCs are divided into three major groups coined ILC1, ILC2, and ILC3. Similar to NK cells, ILC1s promote type 1 immune responses against intracellular pathogens via the production of IFN-γ and TNF. However, ILC1s lack perforin-dependent cytotoxicity but they are able to kill target cells via alternative pathways, such as TNF receptor-mediated induction of apoptosis. ILC1s do not develop in the absence of the TF T-bet, which is evolutionarily related to Eomes.6,8 ILC2s depend on the lineage-specifying TF GATA-3 and secrete classical type 2 effector cytokines, such as interleukin (IL)-4, IL-5, IL-9, and IL-13, as well as amphiregulin (AREG), which promote type 2 inflammation in the context of anti-helminth immunity, allergic reactions or tissue remodeling.9,10,11,12,13,14 ILC3s comprise of different populations of retinoic acid receptor-related orphan receptor (ROR)γt-dependent lymphocytes, which secrete IL-22 to fortify epithelial barriers.15,16,17 These include fetal lymphoid-tissue inducer (LTi) cells,18 which are essential for the formation of secondary lymphoid tissues during development. In adult mice, CCR6+ ILC3s have a similar phenotype to LTi cells and are therefore referred to as LTi-like lymphocytes. In the intestine, they are located in cryptopatches and produce IL-17 in addition to IL-22.19,20,21 The second subset of ILC3s in adult mice lacks CCR6 expression (CCR6− ILC3s) but co-expresses the TF T-bet in addition to RORγt. Upregulation of T-bet is establishing a transcriptional program in these cells, which is similar to one of ILC1s (ILC1-like) characterized by loss of the lineage-specifying TF RORγt and upregulation of natural killer-cell receptors and markers of type 1 immunity, such as NKp46, NKG2D, NK1.1, and IFN-γ.21,22,23,24,25
NK cells have a similar migratory pattern to T and B cells and express the adhesion molecule CD62L, which allows them to transmigrate through high endothelial venules (HEVs) from the bloodstream into lymphoid structures.6 As a consequence, NK cells are well represented in secondary lymphoid organs, whereas other ILC subsets, which lack CD62L are predominantly tissue-resident lymphocytes.26 As tissue-resident cells, ILCs integrate into the organ milieu and only circulate in the bloodstream in low numbers but are often replenished by local precursors.27,28,29 This notion is supported by parabiosis experiments where mainly donor-derived adaptive lymphocytes and NK cells, but not other ILCs subsets could be recovered from the recipient’s tissues at steady state.26,30,31 During worm infection, ILC2s and in particular inflammatory ILC2s can leave their environment and are disseminated to other tissues via the bloodstream.32,33,34 Because of their localization at barrier surfaces, which are often exposed to pathogens, allergen or irritants, ILCs are first responders during immune activation and thereby influence the cytokine milieu and the adaptive immune response. Uncovering how ILCs are regulated and how they interact with tissue-resident cells to maintain organ homeostasis is key to understand how protective or detrimental immune responses in tissues are generated.
ILC development
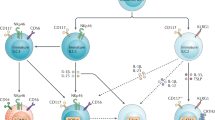
In the bone marrow, hematopoietic stem cells are the origin of the two major hematopoietic branches, the myeloid and the lymphoid lineage represented by the common myeloid precursor (CMP) and common lymphoid precursor (CLP), respectively.35,36 The CLP is phenotypically defined as CD127+ (IL-7Rα+) Flt3+ c-Kitint Sca-1int cells among lineage-negative (Lin−) cells, illustrating the importance of IL-7 for the development of the lymphoid lineage, and is giving rise to both innate and adaptive lymphocytes.37 Innate and adaptive lymphocyte development diverges after the CLP stage and committed precursor cells could be defined, which still possess a multi-lineage potential for different ILC lineages, but not for adaptive lymphocytes6,38,39 (Fig. 1). The earliest stage of these committed innate precursors, coined early ILC precursor (EILP), is defined by expression of the TF T cell factor 1 (TCF-1) and has the potential to give rise to all ILC subsets.38 Somehow counterintuitive is the low expression of IL-7Rα on the EILP because upstream (CLP) and downstream precursors have high IL-7Rα expression. Nevertheless, it was demonstrated that EILP was upstream of subsequent precursors and did not constitute an alternative route of ILC development.40 The modulation of IL-7Rα during development is not unprecedented during lymphocyte development since double-negative thymocytes have high IL-7Rα expression until it is downregulated at the double-positive stage and then again upregulated in single-positive thymocytes.41 The common helper-like ILC progenitor (CHILP) is defined by a surface phenotype similar to CLP, including IL-7Rα expression but lack of CD93 and Flt3, and expression of the transcriptional repressor ID2.6 The analysis of the Id2-deficient mice revealed that these mice lacked all ILC subsets and had a maturation defect in NK cells.11,42,43 While the CHILP had multi-lineage potential documented in several publications, more heterogeneity was found within the population, e.g., the TF PLZF using multicolor approaches.39 Reporter mice and fate-labeling approaches revealed that although ILCs lacked expression of PLZF, they had a history of PLZF expression suggesting that they were derived from a PLZF+ progenitor cell.44 The PLZF+ progenitor cell had a similar phenotype to the CHILP but lacked the potential to give rise to CCR6+ ILC3s. Several approaches including single-cell sequencing confirmed the existence and potential of multi-lineage ILC precursors and revealed additional markers, such as PD-1, which is highly expressed on PLZF+ ILC precursors (ILCP).45,46 Key TFs for early ILC fate commitment in addition to TCF-1, ID2, and PLZF include NFIL3, TOX, and GATA-3.47,48,49,50,51 In line with their role in early ILC development, gene-deficient mice for one of these TFs exhibit a strong developmental defect in all or most ILC lineages except NK cells and LTi cells in GATA-3-deficient mice. For a more detailed discussion about the differentiation potential and transcriptional regulation of ILC precursors, the reader is referred to recently published reviews that extensively discuss this topic.5,52

The hematopoietic stem cell (HSC) is the source of all hematopoietic cells and give rise to the common myeloid progenitor (CMP) and common lymphoid progenitor (CLP), and CLP has the potential to develop into all lymphocytes. The early ILC progenitor (EILP) gives rise to all ILC subsets, whereas common helper-like ILC progenitor (CHILP) and the ILC progenitor (ILCP) still have multi-lineage potential but this is restricted to ILC1, ILC2, and ILC3 as indicated by arrows. Transcriptional requirements, phenotypical markers, and effector molecules of ILCs are shown (TCR T cell receptor, BCR B cell receptor).
A plethora of TFs was found to either affect development or maturation of NK cells including ETS-1, PU.1, IKAROS, EOMES, ZEB2, AIOLOS, PRDM1 (BLIMP1), FOXO1, IRF2, RUNX3, and KLF2.52 Among them, Eomes appears as relatively selective because it is not expressed in other ILC subsets, but in CD8+ T cells in mice; and conditional gene targeting of Eomes using NKp46Cre resulted in the depletion of NK cells without affecting other innate or adaptive lymphocytes.7,53,54 However, Eomes expression was found in human ILC1s in some tissues and, therefore, is not a selective marker to distinguish NK cells and ILC1s in humans.55 It should be noted that in contrast to ILC1s, NK cells were only mildly affected by the lack of T-bet resulting in altered migration, but unperturbed development of NK cells.56 In contrast, ILC1 development is strictly dependent on T-bet but not on Eomes.6,8,57 The TF Hobit was discovered as an essential regulator for the development of tissue-resident T cells,58 and was later shown to be essential for the development of liver ILC1s but not ILC1s in other organs.59
The development of ILC2s depends on the TF GATA-3, BCL11b, RORα, and Gfi1.9,14,60,61,62 GATA-3 is considered as lineage-specifying TF although it is required for the development of multiple ILC lineages because GATA-3 is an essential regulator of early ILC development on the CHILP level. Hence, GATA-3 is similarly active as in T cells, in which GATA-3 regulates not only T helper (Th)2 fate decision but also several steps during development including early lineage commitment.47,63
The development of all ILC3 subsets strictly depends on the lineage-specifying TF RORγt.19,64,65 CCR6+ LTi-like ILC3s are developmentally affected in TOX-, NFIL3-, or GATA-3-deficient mice,48,51,66 and these three TFs mediate their function in early ILC development. While CCR6+ ILC3s developed in mice deficient in the aryl hydrocarbon receptor (AHR), they were functionally impaired, illustrated by the altered maturation of cryptopatches in Ahr−/− mice, whereas postnatally emerging CCR6− ILC3s did not develop.21,67,68 CCR6− ILC3s develop after birth and co-express T-bet, which is competing with RORγt in determining the cell fate of ILC3s21,22,23 and which is establishing a type 1 program accompanied by a loss of RORγt and IL-22 production and upregulation of NK receptors and T-bet target genes, such as NKp46, NKG2D, and NK1.1, IL12Rβ2, CXCR3, and IFN-γ.24,25 In the absence of T-bet, CCR6– ILC3s still developed but failed to induce the type 1 program. Notably, T-bet-deficient ILC3s could still trigger colitis through secretion of IL-17A.21,69 The balance between RORγt and T-bet is regulated by the TF c-Maf, which repressed T-bet expression by binding to the promoter and therefore suppressing the type 1 program in ILC3s.70,71
NK cells
Immune recognition strategies used by NK cells
Immune recognition in both the innate and adaptive immune system relies to a large extent on the interaction between immunoreceptors and the corresponding ligands. Moreover, such immunoreceptors in most cases detect non-self peptides with respect to the T cell receptor (TCR) or B cell receptor (BCR), or ligands recognizing a broad biochemical spectrum in case of pattern recognition receptors.72 While NK-cell-mediated immune surveillance is similarly dominated by receptor-ligand interaction, immune recognition strategies used by NK cells exceed non-self recognition and are mainly based on recognition of self-molecules, coined missing-self and induced-self recognition (Fig. 2).73 Notably, the immune regulation of NK cells by missing-self and induced-self ligands is very dominant resulting in altered NK development when missing-self and induced-self ligands are absent.74,75 Immune recognition strategies of NK cells have similarities to T cells, because non-self, missing-self, and induced-self either recognizes classical MHC I (missing-self) or non-classical MHC I molecules in case of some non-self and induced-self ligands. Missing-self recognition is based on inhibitory receptors of the Ly49 (mouse) or killer-cell immunoglobulin-like receptor (KIR, human) family expressed on NK cells, which detect classical MHC I on target cells resulting in their lysis in the absence of MHC I.76 Concerning non-self recognition, non-classical MHC I molecules Qa-1/HLA-E present foreign peptides derived from murine cytomegalovirus (MCMV) to the NKG2C/CD94 receptor expressed on NK cells thus forming a complementary innate receptor-ligand pair to the peptide MHC-TCR interaction in the adaptive immune system.77 In contrast, ligands for the stimulatory immunoreceptor NKG2D are non-classical MHC I molecules that do not present peptides and that are not expressed by healthy cells, but that are upregulated upon infection or transformation in tumor cells.78 Likewise, B7-H6, which has structural similarities to the B7 co-stimulatory molecules, was induced on tumor cells and stimulated anti-tumor immunity via binding to NKp30.79 NKp30 belongs together with NKp44 and NKp46 to the family of natural cytotoxicity receptors.80 NKp44 recognized PDGF-DD81 and thereby acted as a factor, which promoted tumor growth, whereas NKp46 bound complement factor P (properdin) and was involved in the control of Neisseria meningitides infections.82 Likewise, NK cells recognize the fragment crystallizable (Fc) portion of antibody via the Fc receptor CD16 and lysed antibody-coated cells by antibody-dependent cellular cytotoxicity (ADCC). NK cells integrate stimulatory or inhibitory signals from self-ligands, including but not limited to Tigit, DNAM-1, 2B4, and PD-1, which define the activation threshold or cell adhesion of NK cells.5,52

NK cells are regulated by recognition of non-self, missing-self, and induced-self ligands. Receptor-ligand interactions and factors regulating NK-cell activation as well as effector functions are shown. MNP mononuclear phagocyte, DC dendritic cell, GR glucocorticoid receptor (Nr3c1), ADCC antibody-dependent cellular cytotoxicity.
In addition to membrane-bound receptor-ligand interaction, NK cells are regulated by humoral factors e.g. cytokines, such as IL-15, IFN-I, IL-27, IL-12, and TGF-β, but also glucocorticoids. IL-15 is essential for the development and activation of NK cells and is often trans-presented via the IL-15Rα-chain expressed by dendritic cells (DCs) to the low-affinity IL-2/IL-15 receptor on NK cells composed of the IL-2Rβ-chain CD122 and the common γ-chain CD132.83 DCs produce additional cytokines, such as IFN-I, IL-27, and IL-12 that are required for priming and activation of NK cells.84 While IL-12 was originally described as an NK-cell-stimulating factor,85 several publications reported its stronger effects on ILC1s or ILC3s than on NK cells.6,59,84,86 Moreover, NK cells were responsive to glucocorticoid signals via expression of the nuclear receptor Nr3c1 (glucocorticoid receptor) and were therefore regulated by neuroendocrine signals from the hypothalamic-pituitary-adrenal axis. Glucocorticoids prevent IFN-γ production by NK cells in conjunction with the inhibitory receptor PD-1 and thus control susceptibility to MCMV infection and sepsis.87,88
In summary, NK cells are patrolling innate lymphocytes that test target cells for the presence and absence of ligands to eliminate the target cell if necessary. Additional cytokine signals, such as IL-15, IFN-I, and IL-27 regulate NK-cell development and activation.
NK cells fight intracellular infections and tumors via cell-mediated cytotoxicity and production of IFN-γ
NK-cell activation is to a large extent regulated by the balance between stimulatory and inhibitory signals received by their receptors. If the activation threshold is exceeded, a response is triggered, which results in the specific lysis of the target cell or secretion of the cytokine IFN-γ. To mediate cytotoxic activity, the cytoskeleton is reorganized toward the target cell, and an immunological synapse is formed resulting in the release of granules that contain perforin and granzymes. Perforin is a pore-forming molecule, which ruptures the plasma membrane of the target cell and granzymes are proteases that induce apoptosis via different mechanisms including cleavage of caspase 3. The target cell is often an infected cell, which is removed via cell-mediated cytotoxicity to control the infection. Elimination of hematopoietic cells via cell-mediated cytotoxicity was described as an immune regulatory mechanism as well, e.g., during infection with lymphocytic choriomeningitis virus (LCMV).89,90 IFN-γ is an equally important effector molecule produced by NK cells since it activates antimicrobial functions in macrophages, increases antigen presentation and immunoglobulin (Ig) class switching. Deficiency in either perforin or IFN-γ production results in susceptibility to infection with MCMV, a viral infection that is largely controlled by NK cells (Table 1).91,92,93 MCMV belongs to the β-herpesvirus family of double-stranded DNA viruses that establish long term persistent infection in the host by manipulating the immune response and in particular MHC I expression and recognition by NK cells via NKG2D ligands. MCMV encodes several proteins that interfere with antigen presentation, of which m157 mimics MHC I, probably to avoid missing-self recognition by NK cells through the engagement of inhibitory Ly49 receptors. Some mouse strains have developed a stimulatory Ly49 receptor called Ly49H to prevent immune evasion by MCMV. Ly49H recognized m157 and dominated the immune responses to MCMV illustrated by superior viral control in mouse strains that carry Ly49H.94
The immunosurveillance of tumors by NK cells is controlled by immunoreceptors, such as NKG2D, NKp30, and NKp44. We will focus here on anti-tumor immunity mediated by NKG2D because it is best understood due to the availability of knockout mouse models. Ligands for the NKG2D receptor were found to be expressed on stressed or transformed cells, but not on healthy cells.95 Ectopic overexpression of NKG2D ligands on tumor cells resulted in the rejection of the tumor cell line by the immune system and even formation of immunological memory.96 Further, NKG2D-deficient mice were more susceptible to epithelial and lymphoid tumors that expressed NKG2D ligands.97 While shedding of NKG2D ligands on the surface of tumor cells was interpreted as a tumor escape mechanism,98 recent data suggest that shedding might prevent the downregulation of NKG2D and therefore promote anti-tumor immunity.99 Clinical trials are undertaken to investigate how this basic knowledge about checkpoints of NK-cell regulation could be translated to trigger anti-tumor immunity in patients.100
ILC1s
The term ILC1 includes several populations of innate lymphocytes that are developmentally distinct from conventional NK cells. ILC1s were described in many tissues including TNF-related apoptosis ligand (TRAIL)+ NK cells/ILC1s in the liver,101 thymic NK cells,102 ILC1s in the intestinal lamina propria6 and epithelium,55 in adipose tissue103 and in the uterus.104 In most tissues and with the exception of thymic NK cells,102 ILC1s developmentally depend on the TF T-bet and the cytokine IL-15 but not on IL-7, although they express IL-7R.6,8 Since ILC1 populations found in different tissues sometimes have a slightly different phenotype or developmental requirements, it is still unclear whether this reflects tissue adaption within one lineage or several lineages of innate lymphocytes. Further work is needed to elucidate these aspects.
Although ILC1s expressed a similar array of killer receptors as NK cells, such as NKp46, NKG2D, and NK1.1, the functional implications of these receptors for ILC1 biology remain understudied (Fig. 3). However, a point mutation in NKp46 was recently identified in CD45.1 mice, which prevented membrane trafficking of the receptor. As a consequence, ILC1s from mice carrying the point mutation in NKp46 had diminished surface expression of TRAIL resulting in decreased anti-tumor immunity.105,106 Moreover, this mutation also affected the function of NK cells and NKp46+ ILC3s, which all express NKp46.105,106

Multiple interactions between ILC1 and hematopoietic or parenchymal cells are depicted. MF macrophage, DC dendritic cell, MCMV murine cytomegalovirus, GR glucocorticoid receptor (Nr3c1).
Ly49 receptors are strongly underrepresented on ILC1s suggesting that ILC1s do not rely on missing-self recognition.6,8 ILC1s were first described in the liver as TRAIL+ NK cells and based on their phenotype and different functional properties they were interpreted as immature NK cells.101 Although TRAIL could be induced on activated classical NK cells, it is highly expressed at steady state on ILC1s. TRAIL can engage DR4/DR5 receptors on the target cell and induce apoptosis through Fas-associated protein with death domain (FADD) as well as caspase-8 and -10, a pathway different from perforin/granzyme-mediated cytotoxicity of NK cells. While this pathway has been linked to anti-tumor immunity, the resistance of cancer cells to TRAIL-mediated apoptosis has also been reported.107
A very intriguing aspect of ILC1 biology is their memory-like property. Although poorly understood on a molecular level, the concept is based on the finding that NKp46+ lymphocytes from the liver (ILC1) but not from the spleen could trigger hapten-specific immune responses leading to skin contact hypersensitivity, which did not require the adaptive immune system.108 ILC1 memory was formed following hapten sensitization of the skin in the skin draining lymph nodes and required IL-7 and CXCR3 signaling. Moreover, the homing of ILC1s to the liver was mediated by CXCR6 on ILC1s and the ligand CXCL16.109,110 The ILC1 memory response was not limited to contact hypersensitivity but was also shown to mount antigen-specific recall responses, which mediated immunity against viruses including vesicular stomatitis virus, influenza and human immunodeficiency virus type 1 (HIV-1).110 While antigen-specific memory responses were linked to ILC1s,108,109 ILC2s were shown to adopt a memory-like phenotype and function after papain challenge or IL-33 exposure in an antigen-unspecific manner.111
Although originally believed to be limited to adaptive lymphocytes, memory-like behavior of ILCs was also reported. However, unlike the molecular machinery regulating adaptive memory responses, the receptors mediating antigen-specific memory of ILC1s require further investigation.
In addition to memory responses, ILC1s can mediate protection against acute infections, such as MCMV infection. Hobit−/− mice, which lack liver ILC1s, were more susceptible to MCMV infection in the liver at early time points. This is a remarkable finding because MCMV is controlled at later stages by NK cell via Ly49H receptors.94 Mechanistically, ILC1s were stimulated by IL-12 derived from conventional dendritic cells to secrete IFN-γ59 and did also mount a memory response driven by the MCMV protein m12.112 ILC1s contributed to host defense against infection by secretion of IFN-γ and TNF in mouse models of Clostridium difficile and Toxoplasma gondii infection, but were also implicated in the development of chronic or excessive inflammation during colitis, multiple sclerosis, and sepsis.6,8,53,55,87,113 The effect of ILC1-derived IFN-γ following IL-12 release from myeloid cells was not limited to infection, but also regulated macrophage polarization towards an M1 phenotype, which promotes obesity and insulin resistance.103
Protective effects of ILC1s were described during sterile inflammation in the liver after injection of carbon tetrachloride which triggered acute liver injury. ILC1s were activated via DNAM-1 and the ligand CD155 expressed on hepatocytes together with the release of IL-7 and ATP, which stimulate IFN-γ production from ILC1s. IFN-γ induced survival signals in hepatocytes mediated by the anti-apoptotic protein Bcl-XL.114
Although development and regulation of NK cells and ILC1s are distinct, the overlap in effector functions, such as IFN-γ and promotion of type 1 immunity together with the limitations in specifically targeting these subsets has thus far hindered dissection of the unique functions of ILC1s, and further research will be required to address this question.
ILC2s
Regulation of ILC2s
Immune recognition strategies of the innate immune cells, such as myeloid cells are largely dominated by pattern recognition receptors for a big variety of non-self molecules and presentation of non-self peptides to adaptive lymphocytes via MHC. Upon pathogenic encounter, myeloid cells secrete cytokines and migrate to secondary lymphoid organs to present non-self peptides to adaptive lymphocytes.72 This provokes the question of whether ILCs have similar properties to their innate myeloid counterpart in terms of sensing via pattern recognition receptors and antigen presentation.
While reports about pattern recognition receptors on helper-like ILCs are scarce, presentation of peptide-MHC was found to regulate adaptive immune response as discussed in section ‘Regulation of adaptive immune responses by ILCs’ in this review. Although membrane-bound receptor-ligand interactions regulate helper-like ILC functions, immune receptor-ligand interaction appears less dominant in regulating these cell subsets compared with soluble factors. This is also in line with the concept of tissue-resident cells, which are embedded into the fabric of tissues and require diffusible effectors as opposed to patrolling immune cells.
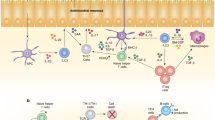
ILC2s are mainly regulated by soluble factors including cytokines, neuronal factors, inflammatory mediators, and hormones. In addition to IL-2 and IL-7, which are broadly sensed by ILCs and adaptive lymphocytes, the cytokines IL-4, TNF-like ligand 1 A (TL1A), transforming growth factor (TGF)-β, stem cell factor (SCF) stimulate ILC2 activation and the alarmins IL-25, IL-33, and thymic stromal lymphopoietin (TSLP) are major activators of ILC2s (Fig. 4).10,11,12,115,116,117 IL-25 is secreted by specialized epithelial cells called tuft cells, which express chemosensory receptors detecting succinate production by worms and protozoans.118,119,120,121,122 IL-25 is released following helminth infections and triggers activation and generation of a subset of ILC2s coined inflammatory ILC2s. Inflammatory ILC2s promote anti-helminth immunity and were described in the mesenteric lymph nodes and lungs following worm infection but were barely detectable at steady state in these organs.32,33 However, intestinal ILC2s, which express high amounts of the IL-25-receptor component IL-17RB and low amounts of the IL-33-receptor subunit ST2, are phenotypically similar to inflammatory ILC2s. Therefore, these cells were proposed to differentiate into inflammatory ILC2s and migrate to the mesenteric lymph nodes upon worm infection.4,32,33,120,123 Generation of inflammatory ILC2s was promoted by the enzyme tryptophan hydroxylase 1, which was required for anti-helminth immunity and which was induced in ILC2s following worm infection or IL-33 treatment.123 In contrast to IL-25, recent data revealed that IL-33 was expressed in PDGFRα+ stromal cell and pre-adipocytes in different tissues including the adipose tissue, lung and intestine.123,124,125,126,127 Finally, adventitial stromal cells express IL-33 and are a major source of TSLP. These data indicate that non-hematopoietic cells, such as Tuft cells or adventitial stromal cells might be signaling hubs in tissues that are specialized in triggering type 2 immune responses. ILC2 activation is further regulated by inflammatory mediators of the arachnoid acid pathway, such as prostaglandin (PG) D2 and leukotriene D4 via CRTH2 and CysLTR1, respectively. In contrast, PGE2 and lipoxin A4 (LX4) inhibited ILC2 activation via EP4 and ALX receptors, respectively.128,129,130,131 Additional negative regulators of ILC2 function include cytokines that promote type 1 immune responses,31,132,133 such as type I and II interferons, as well as IL-27 but also glucocorticoids,87,88 male sex hormones134 and some metabolites. Interestingly, the dietary metabolite retinoic acid contained in carrots and the phytochemical indole-3-carbinol, which is an AHR ligand found in cruciferous vegetables, such as cabbage and broccoli, restrain ILC2 responses, whereas they stimulate ILC3s. Consequently, dietary metabolites can shape the immune responses via regulation of ILC2s and ILC3s as demonstrated during helminth and Citrobacter rodentium (C. rodentium) infections.135,136,137,138

ILC2s maintain interactions between neurons, epithelial cells, stromal cells, adipocytes but also myeloid cells and adaptive lymphocytes to regulate tissue homeostasis. Baso basophil granulocyte, Eos eosinophil granulocyte, MF macrophage, DC dendritic cell, SC stromal cell, AAM alternatively activated macrophage, AR androgen receptor, AHR aryl hydrocarbon receptor, Arnt nuclear translocator of AHR, RAR retinoic acid receptor, RXR retinoid X receptor, Arg1 arginase, IgA immunoglobulin A, EC epithelial cell, GC goblet cell, TC tuft cell.
ILC2s are phenotypically defined by expression of the inhibitory receptor KLRG1, which recognizes E- and N-cadherins, and which could therefore transduce inhibitory signals upon ligand binding. However, although inhibitory effects of KLRG1 on human ILC2s were reported in vitro, the in vivo relevance remains to be demonstrated. In this context, it should be noted that the genetic ablation of Klrg1 did not alter T cell activation in vivo, nor did it lead to hyperreactive T cells.139,140 Further, ILC2s provide co-stimulatory signals to each other using inducible T cell co-stimulator ligand (ICOSL) and glucocorticoid-induced TNFR-related ligand (GITRL) combined with autocrine or paracrine secretion of IL-9.133,141,142,143,144 ILC2s are repressed via PD-1 that is expressed on activated ILC2s, and blocking or genetic deletion of PD-1 results in exaggerated type 2 inflammation promoting allergic asthma or worm resistance.45,145 ILC2s maintain multiple interactions in tissues with both hematopoietic and non-hematopoietic cells to regulate type 2 inflammation (Fig. 4). Among the strongest stimulators of ILC2s are alarmins, which are secreted by specialized epithelial cells and stromal cells.
Effector functions of ILC2s
ILC2s mediate their function mainly by secretion of soluble effector molecules, in particular classical type 2 cytokines IL-5, IL-9, and IL-13 and under some conditions IL-4.10,11,12,144,146 In general, these cytokines promote type 2 inflammation by acting on diverse cell types. IL-9 is mainly produced by ILC2s in an autocrine or paracrine manner thus leading to the expansion of ILC2s, which also carry the IL-9 receptor. IL-9-dependent expansion of ILC2s was shown to be crucial for tissue repair following lung inflammation or resolution of arthritis.143,144,147 IL-5 is important for activating innate myeloid cells, which are involved in type 2 immunity. In particular, IL-5 mediates recruitment and activation of eosinophils, which secrete IL-4, further promoting type 2 inflammation during allergic airway inflammation but also metabolic homeostasis.148 While IL-13 promotes type 2 inflammation via differentiation of alternatively activated macrophages (AAMs), it is the direct effector cytokine promoting worm expulsion via stimulating smooth muscle contraction and hyperplasia of goblet cells resulting in enhanced mucus production as well as IL-25 secretion from tuft cells to further boost type 2 inflammation.3 IL-25 is a strong inducer of ILC2 activation and inflammatory ILC2s. Such stimuli promote migration of ILC2s from their tissue of origin to the blood and dissemination to other tissues where they promote systemic type 2 inflammation.32,33,34
It is well established that type 2 immunity promotes expulsion of helminth parasites and noxious environmental substances as well as tissue repair.149 Since the crucial cytokine IL-13 is secreted by both Th2 cells and ILC2s, it raises the question about the contribution of both cell types to the immune response against helminths. Published evidence suggests a pivotal role of ILC2s in immunity against Nippostrongylus brasiliensis (N. brasiliensis),10,11 Strongyloides venezuelensis,150 and Trichuris muris151 infections but we will focus the discussion on N. brasiliensis since most data are available from this infection model. While Rag−/− mice have comparable worm counts as wild-type mice at early time points following N. brasiliensis infection, worm expulsion is delayed at day 10 post infection. These data suggest that T cells are dispensable during the early infection phase but mediate worm expulsion at later time points or during reinfection.10,152 It was also demonstrated that injection of recombinant IL-25 or IL-33 promoted worm expulsion in the absence of T cells.10,12,153 Furthermore, Oliphant et al. depleted ILC2s without affecting T cell numbers and reported a higher worm burden in ILC2-depleted mice.154 Although these data argue for a pivotal role of ILC2s also in T cell replete mice during N. brasiliensis infection, anti-helminth immunity is based on a multilayer system involving several cell types including ILC2s and T cells.
Excessive type 2 immune responses can become detrimental and constitute the underlying mechanism in the pathogenesis of atopic disease including allergic asthma, atopic dermatitis, and allergic rhinitis. Increased ILC2 numbers were found in bronchoalveolar lavage (BAL) of asthmatic patients,155 in nasal polyps of patients with chronic rhinosinusitis13 and skin lesions of atopic dermatitis patients.140,156 In mouse models, multiple studies suggest a pivotal role for ILC2s in the pathogenesis of allergic asthma provoked by the inhalation of the protease papain, the fungal extract from Alternaria alternata or in airway hyperreactivity following influenza virus infection.31,157,158 Further, depletion of ILC2s in the papain model resulted in decreased Th2 cell numbers and type 2 inflammation. Moreover, depletion of ILC2s after the primary challenge with papain revealed defects in memory Th2 cell formation mediated by ILC2s and myeloid cells.159,160
ILC2 function was investigated in a mouse model of atopic dermatitis provoked by the vitamin D3 analog calcipotriol. While several studies have revealed that ILC2s promote atopic dermatitis, ILC2 activation in the skin might be in parts differentially regulated since ILC2s responded to IL-18, whereas the role of IL-33 in stimulating ILC2s appeared to be context dependent.4,140,156,161
Since the life cycle of helminths, such as N. brasiliensis creates tissue damage, e.g., the rupture of capillaries in the lungs, type 2 immune responses have not only developed to combat helminth infection but also to promote tissue repair and remodeling through secretion of molecules, such as AREG.147,162 AREG was secreted by ILC2s and acted as a growth factor that binds to the EGF receptors expressed on epithelial cells and promoted cell proliferation and tissue repair following influenza virus infection in the lung or during dextran sulfate sodium (DSS)-induced colitis.163,164
ILC2s contribute to acute inflammation in the context of hepatitis.165 However, ILC2s were found to mediate important functions in the resolution of chronic inflammation during arthritis by induction of regulatory T cells via ICOSL and GITRL.143 Moreover, multiple studies support a role for ILC2s in wound healing and tissue remodeling after infection or tissue damage in the skin, lung, and intestine.147,163,164,166 In addition, ILC2s also stimulated irreversible changes in the organ structure as they were implicated in liver fibrosis development.167
While limitations in specifically targeting ILC2s in the presence of T cells have to be considered when drawing conclusions from disease models, the experimental evidence for a role of ILC2s in promoting type 2 inflammation in the context of allergic asthma, atopic dermatitis, and helminth infections are encouraging and could provide novel targets for pharmacological intervention.
ILC3s
Formation of lymphoid organs and interaction in cryptopatches
In the fetal period, LTi cells are essential for the formation of lymphoid organs, such as lymph nodes and Peyer’s patches. Following embryonic day 12.5, CXCR5+ LTi cells are clustering and form lymph node anlagen together with mesenchymal stromal cells. Formation of lymph node anlagen is promoted by retinoic acid and CXCL13 secretion thus attracting LTi cells. It was proposed that neuronal stimuli might trigger the initial steps in the formation of lymph node anlagen but this model is not yet supported by strong evidence.168 Factors promoting lymph organogenesis include IL-7, SCF, TSLP, TRANCE (RANKL), and LIGHT leading to the expression of LTα1β2 on ILC3s, which binds LTβ receptor on the mesenchymal cells, a decisive event in the formation of lymphoid organs (Fig. 5).137,168 The interaction results in further secretion of the chemokines CXCL13, CCL19, and CCL21, which attract adaptive lymphocytes to the lymph node anlagen and in the upregulation of adhesion molecules VCAM-1, ICAM-1, and Madcam-1, which facilitate the formation of lymphoid organs.168 This process is largely controlled by cell migration and the interaction of cell surface molecules during fetal development. Some of the molecular machinery is also maintained in CCR6+ ILC3s, the adult counterpart of LTi cells, which are located in cryptopatches in the intestine. CCR6+ ILC3s were attracted to the cryptopatches via the chemokine CCL20. Cryptopatch formation is altered in the absence of AHR or c-Kit-SCF signaling, which is controlled by AHR.68,136 Within cryptopatches, CCR6+ ILC3s interacted with myeloid cells via LTα1β2-LTβ receptor to stimulate secretion of IL-23, which resulted in increased IL-22 secretion and enhanced production of antibodies in cryptopatches.169,170,171 Cryptopatches were found in close proximity to nerve fibers, which were surrounded by glial cells. Glial cells secreted glial cell-derived neurotrophic factor (GDNF), which stimulated IL-22 production by ILC3s via the cell surface receptor Ret, and which provided protection in the context of C. rodentium infection and DSS-induced colitis.172

Regulatory circuits that involve ILC3s include interaction with myeloid cells, epithelial cells, stromal cells, neurons, and adaptive lymphocytes. Although depicted as one cell, it should be noted that CCR6+ ILC3s and CCR6− ILC3s are developmentally and functionally distinct. MNP mononuclear phagocyte, 7α,25 OHC 7α25-hydroxycholesterol, M macrophage, SC stromal cell, EC epithelial cell, ISC intestinal stem cell, AHR aryl hydrocarbon receptor, Arnt nuclear translocator of AHR, RAR retinoic acid receptor, RXR retinoid X receptor, SCFA short chain fatty acid.
Regulation of ILC3s
Similar to other ILCs, ILC3 activation is mainly regulated by soluble factors, such as cytokines, neuronal factors, metabolites, and inflammatory mediators. Important regulatory cytokines include IL-1α, IL-1β, IL-2, IL-7, IL-23, TL1A, SCF, and TSLP, which are secreted by a variety of cells including myeloid cells, T cells, epithelial cells, and stromal cells.3 ILC3s do not develop in the absence of IL-7, which is important for the development of most innate and adaptive lymphocytes. ILC3s produce IL-2 but also compete with T cells for this cytokine.173,174 IL-1β, IL-23, and TL1A are mainly produced by mononuclear phagocytes (MNPs), which are closely entangled with ILC3s, and which are potent stimulators of IL-22 production and ILC3 activation.175,176 TL1A induces expression of OX40L on ILC3s thereby promotes co-stimulation of T cells during colitis.177 Epithelial IL-1α was shown to promote IL-22 production by ILC3s following rotavirus infection.178
While the crucial role for retinoic acid in formation of lymph organs was already discussed in the paragraph above, retinoic acid is also involved in the regulation of gut homing receptors by inducing a switch from CCR7 to CCR9 in ILC3s and ILC1s in gut-associated lymphoid tissue.179 Other molecules determining ILC3 migration include CXCR6 and G-coupled protein receptor GPR183, which sense CXCL16 and 7α25-hydroxycholesterol, respectively.180,181,182 As opposed to ILC2s, deficiency in retinoic acid causes diminished IL-22 production of ILC3s and susceptibility to C. rodentium infections.135 Similar to the role of AHR ligands in regulating the formation of postnatal lymphoid tissues, AHR ligands are crucial for IL-22 production and Ahr−/− mice succumbed to C. rodentium infection.67 Further, ILC3s were stimulated by short chain fatty acid, via the FFAR2 receptor, which promoted IL-22 production and protection from colitis.183 Another study also identified prostaglandin E2 as an inflammatory mediator to prevent the systemic spread of bacteria by promoting barrier functions via the EP4 receptor on ILC3s and enhanced IL-22 production.184
In summary, ILC3 activation is regulated to a large extent by humoral factors released by myeloid cells and parenchymal cells in the tissue.
Effector functions of ILC3s
IL-22 is the main effector cytokine secreted by both CCR6+ ILC3s and CCR6− ILC3s. Although CCR6− ILC3s produced less IL-22 than CCR6+ ILC3s, they remain to be an important source of IL-22.21 Unlike most interleukin receptors, the IL-22 receptor is mainly expressed on non-hematopoietic cells, especially epithelial cells.185 IL-22 maintained barrier integrity and the containment of intestinal bacteria at steady state and was shown to be protective in various infection models, including C. rodentium, C. difficile, Rotavirus, and Salmonella Typhimurium (S. Typhimurium) infection.16,113,178,186,187 C. rodentium infection is a model for attaching-and-effacing Escherichia coli (E. coli), which is controlled by IL-22.16 While mice deficient in IL-22 signaling were more susceptible to C. rodentium infection, the contribution of ILC3-derived IL-22 to the phenotype is still debated because CD4+ T cells, γδ T cells, and NKT cells are also potential sources of IL-22 in this model. ILC3s were found to be the major source of IL-22 in the first week of C. rodentium infection20,187 and were stimulated via IL-23 derived from Notch-2-dependent DCs or CX3CR1+ MNPs.176,188 Further studies suggest that CCR6+ CD4+ ILC3s are the main producers of IL-22, because depletion using an anti-CD4 antibody resulted in susceptibility to C. rodentium infection, whereas genetic depletion of NKp46+ ILC3s did not affect C. rodentium control on a T cell replete background.187,189,190 Altogether the studies argue for a role of T cells and ILC3s in controlling C. rodentium infection, however, a genetic model that selectively depletes all ILC3s without affecting other cell types is lacking.
IL-22 mediates downstream effects on different types of epithelial cells, such as Paneth cells, intestinal stem cells, and enterocytes via engagement of IL-22Rα1 and IL-10Rβ2 chains triggering a signaling cascade resulting in phosphorylation of STAT3.191 Activation of intracellular signaling resulted in secretion of antimicrobial peptides, such as Reg3β, Reg3γ, S108a, and S109a from Paneth cells. Furthermore, IL-22 promoted barrier functions in enterocytes, which limited the dissemination of bacteria. Together with LTα, IL-22 stimulated the glycosylation of epithelial cells that is crucial for protection against S. Typhimurium186 infection and IL-22 and interferon λ jointly stimulated IL-1α release during Rotavirus infection.16,178,191 IL-22 production protected intestinal stem cells from damage caused by chemotherapy, irradiation or genotoxic stress by metabolites, such as toxic AHR ligands, which could in turn modulate IL-22 production.192,193,194
Both tumor-protective and tumor-promoting effects of IL-22 were reported. IL-22 reduced inflammation and intestinal cancer in the acute phase of DSS-induced colitis by fortifying the epithelial barrier function and limiting the microbial influx. In the recovery phase, IL-22 was found to regulate epithelial proliferation.17,192,195 IL-22 limited the progression from epithelial damage to cancer transformation during chronic inflammation via induction of the DNA damage response in intestinal stem cells in a colorectal cancer model induced by DSS and azoxymethane.192 Tumor-promoting effects of IL-22 were found in adenomatous polyposis coli (APC)Min/+ mice, which develop colorectal cancer due to a mutation in the tumor suppressor gene APC.195 In this model, IL-22 promoted proliferation of epithelial cells and, therefore the availability of bioactive IL-22 controls cancer progression. Hence, mice deficient in IL-22-binding protein, which serves as a decoy receptor for IL-22, had more tumors, whereas Il22−/− mice developed fewer tumors.195
Patients with mutations in RORC had selective immunodeficiency to Candida albicans (C. albicans) and Mycobacterium tuberculosis infections due to a reduction in IL-22 and IL-17A.196 While different cell populations that express RORC probably contribute to this drastic phenotype, ILC3s and especially IL-17A+ ILC3s, which were lacking in these patients, might be of relevance.197 IL-17A is mainly produced by CCR6+ ILC3s, consistent with T-bet as a negative regulator of IL-17A.21 While ILC3s were described as pivotal in the control of oral C. albicans infection, they could also trigger detrimental immune responses.198 These experiments were mainly carried out on a Rag−/− background, where ILC3s might be overactivated due to the lack of adaptive lymphocytes. In Tbx21−/− (T-bet) Rag−/− mice, IL-17A+ ILC3s were shown to drive Helicobacter typhlonius triggered colitis.69 IL-17A derived from CCR6+ ILC3s was identified to be important for airway hyperactivity in the context of high-fat diet (HFD)-induced asthma, in which ILC3s were stimulated by IL-1β released from MNP in a Nlrp3 inflammasome-dependent manner.199
T-bet upregulation in CCR6− ILC3s endows these cells with the functional properties of ILC1s including the production of IFN-γ. IFN-γ derived from ILC3s was shown to mediate both protective immunity against Yersinia enterocolitica (Y. enterocolitica) and Clostridium difficile (C. difficile) infection but could also trigger detrimental immune responses.113,200 Interestingly, during Y. enterocolitica infection ILC3s were activated via herpesvirus entry mediator (HVEM), a member of TNF receptor superfamily and soluble LIGHT.200 However, these cells were also shown to be activated by IL-12 and IL-23 and promote colitis in different models of intestinal inflammation including anti-CD40-induced colitis, Helicobacter Hepaticus-induced colitis, as well as immunopathology in the caecum induced by S. Typhimurium and C. rodentium.21,24,189,190,201
ILC3s modulate myeloid cell activation via secretion of GM-CSF and in this way indirectly regulate adaptive immune responses. Additionally, upon activation of IL-1β via MNP, ILC3s are a crucial source of GM-CSF which promotes Treg cells and in this way mediates oral tolerance in the intestine.202 Moreover, ILC3-derived GM-CSF was shown to attract neutrophils to promote antibody production by marginal zone B cells.203
Regulation of adaptive immune responses by ILCs
Unlike MHC I expression, which is present on almost every cell, expression of MHC II is considered as a hallmark of professional antigen-presenting cells. In parallel to antigen-presenting cells, ILC2s and ILC3s are also equipped with the machinery to process and present peptides on MHC II molecules and are therefore able to directly interact with T cells via MHC II-peptide-TCR complex.154,204 Expression of co-stimulatory molecules on ILCs, such as ICOSL, GITRL, and PD-1L was also reported to provide a second signal for T cell activation. Furthermore, IL-2 and GM-CSF secreted by ILC3s, in a myeloid-derived IL-1β-dependent manner, were found to be important for Treg cell-mediated tolerance induction.174,202 ILCs and T cells both express the high-affinity IL-2 receptor alpha-chain, CD25, which brings them in the position to compete for this essential growth factor, thus adding an additional layer of complexity to the regulation of T cell by ILCs.173
At steady state, CCR6+ ILC3s in the intestine present peptides on MHC II without providing co-stimulation, thus resulting in clonal deletion of the antigen-specific T cells, coined intestinal selection of T cells. Mice with deletion of MHC II in ILC3 developed intestinal inflammation, arguing that the intestinal selection of T cells is necessary to maintain immune homeostasis at mucosal sites.173,204 Further studies discovered that ILC3s upregulated co-stimulatory molecules such as OX40L after stimulation with TL1A or IL-1β during inflammation and thus promoted colitis via activation of pathogenic T cells.177,205
CCR6+ ILC3s are mainly positioned in cryptopatches in the adult intestine, which recruit B cells into the structure and thereby further mature. They are then referred to as isolated lymphoid follicles (ILFs) or mature ILFs.19 ILFs provide a structure for ILC3-B cell interaction and thus lead to a T-cell-independent stimulation of IgA, which has also been observed near the marginal zone in the human spleen.171,203 While IgA production in cryptopatches is considered to be T-cell-independent, it was reported that CCR6+ ILC3s modulate the interaction of follicular T helper cells and B cells via MHC II in the colon-draining lymph node and thus regulate the production of T-cell-dependent high-affinity IgA.206 Further, it was discovered that ILC3s support T-cell-dependent IgA via soluble LTα3 and T-cell-independent IgA via surface LTα1β2.170 Therefore, ILC3s can regulate Ig production via direct and indirect mechanisms.
Consistent with the expression of co-stimulatory molecules on their surface, ILC2s stimulate T cells via MHC II-TCR interaction. Stimulation of Th2 cells by ILC2s promoted IL-2 secretion by Th2 cells, which in turn activates ILC2s to produce IL-13 and promote resistance against worm infection.154 Interestingly, ILC2s could co-stimulate Th2 cells via PD-L1-PD-1 interaction and promote type 2 immunity.207 Moreover, IL-9-dependent expansion of ILC2s was found to be important for the induction of regulatory T cells via ICOSL and GITRL and resolution of chronic inflammation during arthritis.143 Taken together, these studies identify ILCs as pivotal regulators of adaptive lymphocytes that can either promote or dampen the adaptive immune responses depending on the environmental context.
Regulation of ILC responses by the nervous system and by the circadian rhythm
The nervous system and the immune system are composed of complex sensory and effector structures, which continuously monitor homeostasis in tissues by measuring different parameters. Chemo-, mechano-, noci-, thermo-, and photo sensation prevail in the nervous system, whereas the immune system relies on non-self and self-recognition and to a lesser extent chemosensation.208,209 The effector molecules of the nervous system include neuropeptides and neurotransmitters, which transmit information over a short distance. In contrast, immune responses are mainly amplified by humoral factors, such as cytokines and direct cell-to-cell interaction. Functional specialization requires the constant exchange of information to efficiently coordinate responses between the immune and nervous system. In this context, recent research has revealed some of the pathways that define the crosstalk between ILCs and the nervous system.
The enteric nervous system is the largest accumulation of neurons outside the central nervous system and enteric neurons are closely entangled with ILCs.208,209 The crosstalk between neurons and the immune system is bidirectional since neurons express but also sense cytokines, such as TSLP, IL-4, and IL-31, which are known to boost type 2 inflammation.210,211 Furthermore, ILC2s produce neuropeptides, e.g., CGRP, which can also be released by neurons or pulmonary neuroendocrine cells (PNECs) and regulates lung inflammation and immunity against helminths. In addition to producing CGRP, ILC2s are also equipped with the receptor Ramp1/CALCRL to sense CGRP. CGRP triggered inhibitory signals in ILC2s and thus limited ILC2 activation in mouse models of helminth infection or allergic asthma.212,213,214,215 Additional neurotransmitters inhibiting ILC2 activation inculde the signature neurotransmitter of the sympathetic and parasympathetic nervous system norepinephrine and acetylcholine. These two neurotransmitters limit ILC2s via α7 nicotinic acetylcholine receptors and β2-adrenergic receptors216,217 and regulate allergic lung inflammation and worm resistance. Furthermore, acetylcholine signals from the vagal nerve stimulated ILC3s to secrete the immunoresolvent PCTR1 during peritoneal E. coli infections.218 In contrast, the neuropeptides neuromedin U (NMU) and vasoactive intestinal peptide (VIP) are expressed in the enteric nervous system and stimulate ILC2s via Nmur1 and Vipr2, respectively.219,220,221,222,223 NMU is expressed in cholinergic neurons in the intestine. NMU is upregulated following worm infection by excretory-secretory products of worms and regulates worm resistance via Nmur1.221,222 VIP is expressed by nociceptors in the lung and stimulates IL-5 production from several cell types including ILC2s, which act back on neurons to sustain lung inflammation via VIP.220 Similarly, VIP regulates IL-5 production from ILC2s and the number of eosinophils in the intestine and is dependent on circadian oscillations and nutrient intake.219
ILC2 and ILC3 activation in the intestine is controlled by the circadian rhythm, which allows to synchronize immunity and nutrient intake.219,224,225,226,227,228 Circadian regulation of ILC3s probably involves several mechanisms including the regulation of RORγt and Rev-Erbα by the Bmal1:Clock circuit, the regulation by the suprachiasmatic nuclei in the brain and release of the neuropeptide VIP, which is known to be controlled by circadian oscillations.225,226,227,228 Indeed, the deletion of Bmal1 in RORγt+ cells resulted in altered RORγt expression, diminished NKp46+ ILC3s and susceptibility to C. rodentium infection. Furthermore, VIP controlled IL-22 secretion by CCR6+ ILC3s via Vipr2. Seillet and colleagues found that Vipr2-deficient CCR6+ ILC3s produced less IL-22 and exerted reduced protective function in the DSS-induced colitis model.225 However, Talbot and colleagues reported that VIP inhibited CCR6+ ILC3s and conditional targeting of Vipr2 in ILC3s using RORc(γt)Cre Vipr2flox/flox mice resulted in an enhanced antimicrobial function of the intestinal epithelium against segmented filamentous bacteria but reduced lipid uptake.224 Since both stimulatory and inhibitory effects of VIP on CCR6+ ILC3s were observed, the function of this neuropeptide might be context-dependent or the discrepancy between these studies might be explained by differences in the experimental approach or the commensal microbiota. In total, these studies provide evidence that neuronal regulation of ILCs harmonizes immunity at barrier surfaces with the light-dark cycles, nutrient intake and homeostasis.
Cytokine milieu and plasticity of ILC subsets
In parallel to T cell differentiation, where lineage-specifying TFs and downstream signaling mediators of the key cytokine receptors suppress disparate T cell fates, ILCs are negatively regulated by cytokine milieu or lineage-specifying TFs of other ILC subsets. In this context, IL-25 and TSLP were both reported to repress ILC3 activation and IL-22 production in the context of colitis and therefore exacerbate intestinal inflammation.20,229 Moreover, cytokines promoting type 1 immune responses, such as IFN-I, IFN-γ or IL-27 were found to inhibit ILC2s and consequently limit allergic inflammation.31,132 Similar findings were reported for the skin in the context of contact hypersensitivity, in which depletion of ILC2s resulted in an exaggerated type 1 immune response.230
Despite an established transcriptional program defined by a lineage-specifying TF, ILCs are able to adopt alternative cell fates after lineage commitment even at steady state but also in the context of chronic inflammation or a certain microenvironment. Plasticity is driven by down-modulation of the lineage-specifying TFs and induction of a different master TF that is often competing with the original TF in determining the cell fate. Plasticity of ILCs was originally observed in CCR6− ILC3s, which lost expression of RORγt and upregulated T-bet, thus transforming ILC3s in a cell type that phenotypically mirrored ILC1s, such as expression of NKp46, NK1.1, NKG2D, IL12Rβ2, and IFN-γ. Since ex-RORγt+ ILC3s carry receptors for IL-12 and IL-23, they could drive chronic inflammation and immunopathology during colitis in patients with Crohn’s disease or in mouse models of experimental colitis induced by anti-CD40 treatment or infection.21,24,25,189,190,201 T-bet appears as a signaling hub that promoted the loss of RORγt in conjunction with the TF AIOLOS and by integrating diverse signals including Notch, IL-2, and IL-12 that support type 1 cell fate, whereas c-Maf and TGF-β suppress T-bet.21,22,25,70,231 Nevertheless, T-bet-deficient ILC3s were still able to promote detrimental immune response via the production of IL-17A during colitis in response to Helicobacter typhlonius.69 In humans, the differentiation of ILC3s into ILC1s could be reversed by the addition of IL-1β, IL-23 and retinoic acid in vitro.232
Differentiation towards an ILC1-like phenotype is not limited to ILC3s, but was also described for ILC2s and NK cells. Conversion of NK cells into ILC1-like phenotype was accompanied by down-modulation of Eomes and mediated by TGF-β, which is a signature cytokine acting on ILC1.55 ILC1-like NK cells were described in the tumor microenvironment or during cytomegalovirus or T. gondii infection. Notably, ILC1-like NK cells were distinct from both NK cells and ILC1s, were functionally impaired and failed to control tumor growth and cytomegalovirus infection.233,234,235 Plasticity of ILC2s towards ILC1 was described in the lungs of patients suffering from chronic obstructive pulmonary disease (COPD) and was promoted by the cytokines IL-1, IL-12, and IL-18 and connected to the induction of T-bet.236 ILC2 conversion to IL-17-producing ILC3s was reported in mice and humans. In mice, stimulation of ILC2s with IL-25 resulted in inflammatory ILC2s and conversion to IL-17 producing cells, which protect from C. albicans infection.32,237 This process was promoted by Notch signals, which induced upregulation of RORγt and IL-17A in ILC2s. Therefore, these cells were shown to be co-producers of IL-13 and IL-17A thus promoting allergic airway inflammation in the house dust mite model.238 In contrast, in humans, the conversion was promoted by ILC3-stimulating cytokines IL-1β, IL-23 and TGF-β in the context of psoriasis and nasal inflammation.239
ILC2s can switch their cytokine profile to IL-10 production.240,241,242 This process does not involve the upregulation of another lineage-specifying TF but is promoted by different cytokines including IL-2. Furthermore, it was discovered that IL-10 is under transcriptional control of the RUNX/CBF-β complex. Since IL-10 secretion by ILC2s is accompanied by loss of type 2 functional properties, it was also interpreted as exhaustion-like phenotype.242 While several groups accordingly reported IL-10 production by ILC2s under certain conditions,240,241,242 IL-10 was also proposed to constitute a separate regulatory ILC lineage defined by the transcriptional repressor ID3.243 However, since a recent paper published by Bando et al. failed to reproduce the existence of a separate ILC lineage different from known ILC populations, further research is needed to clarify the discrepancy between these findings.241
In summary, some degree of plasticity appears to emerge as a hallmark of ILC subsets and might constitute a mechanism of tissue adaption or immune regulation, however, more detailed in vivo fate mapping studies are required before definitive conclusions can be made regarding the generality of plasticity within ILC populations.
Metabolic homeostasis
Low-grade chronic inflammation is determining energy expenditure in white adipose tissue (WAT), therefore, assigning an important role for the immune system in metabolic hemostasis. Among immune cells, ILCs, and predominantly ILC2s, are well represented in WAT. While type 2 inflammation in WAT is associated with increased energy expenditure, shifting the immune response towards type 1 immunity is associated with increased metabolic risks.244 ILC1s, which accumulated in WAT during obesity, were identified as an important source of IFN-γ. ILC1-derived IFN-γ was stimulated by IL-12 secreted by myeloid cells and supported the differentiation of M1 macrophages promoting obesity and insulin resistance.103
In contrast, ILC2s, whose numbers were decreased during obesity, were demonstrated to promote energy expenditure by diverse mechanisms including recruitment of AAMs.148,245,246 AAM differentiation is promoted by IL-4 mainly secreted by eosinophils but also NKT cells. ILC2s are recruiting eosinophils to WAT via secretion of IL-5.148 Besides the effect on AAMs, IL-4 can also directly act on adipocyte precursors that express the IL-4 receptor and promote differentiation of beige adipocytes.246 Beige adipocytes increase energy expenditure by uncoupling of the electrochemical gradient of the respiratory chain in the mitochondria.244 In addition to type 2 cytokine secretion, ILC2s promoted beiging of WAT by cleavage of the opioid peptide proenkephalin A (Penk) into bioactive methionine-enkephalin via prohormone convertase 1, an enzyme that has previously been associated with obesity.244 Penk stimulated beige adipocytes that expressed the opioid receptor δ1 resulting in upregulation of the uncoupling protein UCP-1, an increase in energy expenditure and a decrease in adipose tissue.245 ILC2 activation is regulated by IL-33 produced from adipocyte progenitors and mesenchymal stromal cells in WAT124,125 and controls UCP-1 in beige adipocytes via activation of type 2 inflammation.247 IL-33 also stimulated ILC2s in pancreatic islets, which then activated DCs to release retinoic acid and promote insulin secretion.248 Furthermore, metabolic homeostasis was linked to IL-22, which can be produced by ILC3s. IL-22 promotes metabolic homeostasis partially via regulating lipid absorption in the intestine, as well as additional mechanisms that require further investigation.224,249
Future perspectives
The last 10 years of ILC research has identified previously unappreciated functional diversity of ILCs. ILCs have been detected in almost every tissue with enrichment at barrier surfaces. ILCs provide not only a first-line defense against infections, but they also have established many local signaling circuits to maintain broad tissue homeostatic functions. While some key signaling pathways mediating tissue homeostasis arise, these need to be further dissected, and additional circuits will likely be revealed. An emerging topic is the functional specialization of non-hematopoietic cells in regulating ILCs. Discovering the pathways utilized by tissue-resident cells to regulate ILC responses and tissue homeostasis has the potential to open up avenues for future research. To investigate these pathways will require new genetic tools to selectively and temporally interfere with parenchymal cells and ILC populations in diverse tissue microenvironments. Notwithstanding that, since ILCs have a similar molecular profile to T cells, one of the major unresolved limitations is still the specific targeting of ILC subsets without affecting adaptive lymphocytes. Given the overlap in effector molecules with T cells, the extent, or lack thereof, of functional redundancy between T cells and ILCs is an ongoing discussion.250 Moreover, future research will likely focus on pivotal immunoregulatory pathways controlled by ILCs that have the potential to be therapeutically harnessed in the context of health and disease.
References
Fan, X. & Rudensky, A. Y. Hallmarks of tissue-resident lymphocytes. Cell 164, 1198–1211 (2016).
Spits, H. et al. Innate lymphoid cells—a proposal for uniform nomenclature. Nat. Rev. Immunol. 13, 145–149 (2013).
Klose, C. S. & Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 17, 765–774 (2016).
Ricardo-Gonzalez, R. R. et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat. Imunol. 19, 1093–1099 (2018).
Vivier, E. et al. Innate lymphoid cells: 10 years on. Cell 174, 1054–1066 (2018).
Klose, C. S. N. et al. Differentiation of Type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157, 340–356 (2014).
Gordon, S. M. et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 36, 55–67 (2012).
Daussy, C. et al. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J. Exp. Med. 211, 563–577 (2014).
Hoyler, T. et al. The transcription factor GATA-3 controls cell fate and maintenance of Type 2 innate lymphoid cells. Immunity 37, 634–648 (2012).
Neill, D. R. et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 (2010).
Moro, K. et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544 (2010).
Price, A. E. et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 107, 11489–11494 (2010).
Mjosberg, J. M. et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 12, 1055–1062 (2011).
Mjosberg, J. et al. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity 37, 649–659 (2012).
Sonnenberg, G. F. et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 336, 1321–1325 (2012).
Zheng, Y. et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14, 282–289 (2008).
Zenewicz, L. A. et al. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29, 947–957 (2008).
Mebius, R. E., Rennert, P. & Weissman, I. L. Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 7, 493–504 (1997).
Eberl, G. et al. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat. Immunol. 5, 64–73 (2004).
Sawa, S. et al. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 12, 320–326 (2011).
Klose, C. S. et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature 494, 261–265 (2013).
Rankin, L. C. et al. The transcription factor T-bet is essential for the development of NKp46(+) innate lymphocytes via the Notch pathway. Nat. Immunol. 14, 389–395 (2013).
Sciume, G. et al. Distinct requirements for T-bet in gut innate lymphoid cells. J. Exp. Med. 209, 2331–2338 (2012).
Vonarbourg, C. et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK Cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity 33, 736–751 (2010).
Bernink, J. H. et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 14, 221–229 (2013).
Gasteiger, G., Fan, X., Dikiy, S., Lee, S. Y. & Rudensky, A. Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350, 981–985 (2015).
Bando, J. K., Liang, H. E. & Locksley, R. M. Identification and distribution of developing innate lymphoid cells in the fetal mouse intestine. Nat. Immunol. 16, 153–160 (2015).
Schneider, C. et al. Tissue-resident group 2 innate lymphoid cells differentiate by layered ontogeny and in situ perinatal priming. Immunity 50, 1425–1438 (2019).
Ghaedi, M. et al. Single-cell analysis of RORalpha tracer mouse lung reveals ILC progenitors and effector ILC2 subsets. J. Exp. Med. https://doi.org/10.1084/jem.20182293 (2020).
Peng, H. et al. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J. Clin. Investig. 123, 1444–1456 (2013).
Moro, K. et al. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat. Immunol. 17, 76–86 (2016).
Huang, Y. et al. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat. Immunol. 16, 161–169 (2015).
Huang, Y. et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science 359, 114–119 (2018).
Ricardo-Gonzalez, R. R. et al. Tissue-specific pathways extrude activated ILC2s to disseminate type 2 immunity. J. Exp. Med. https://doi.org/10.1084/jem.20191172 (2020).
Kondo, M., Weissman, I. L. & Akashi, K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 91, 661–672 (1997).
Akashi, K., Traver, D., Miyamoto, T. & Weissman, I. L. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404, 193–197 (2000).
Schlenner, S. M. et al. Fate mapping reveals separate origins of T cells and myeloid lineages in the thymus. Immunity 32, 426–436 (2010).
Yang, Q. et al. TCF-1 upregulation identifies early innate lymphoid progenitors in the bone marrow. Nat. Immunol. 16, 1044–1050 (2015).
Constantinides, M. G., McDonald, B. D., Verhoef, P. A. & Bendelac, A. A committed precursor to innate lymphoid cells. Nature 508, 397–401 (2014).
Harly, C., Cam, M., Kaye, J. & Bhandoola, A. Development and differentiation of early innate lymphoid progenitors. J. Exp. Med. 215, 249–262 (2018).
Sudo, T. et al. Expression and function of the interleukin 7 receptor in murine lymphocytes. Proc. Natl. Acad. Sci. USA 90, 9125–9129 (1993).
Yokota, Y. et al. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature 397, 702–706 (1999).
Boos, M. D., Yokota, Y., Eberl, G. & Kee, B. L. Mature natural killer cell and lymphoid tissue-inducing cell development requires Id2-mediated suppression of E protein activity. J. Exp. Med. 204, 1119–1130 (2007).
Constantinides, M. G. et al. PLZF expression maps the early stages of ILC1 lineage development. Proc. Natl. Acad. Sci. USA 112, 5123–5128 (2015).
Yu, Y. et al. Single-cell RNA-seq identifies a PD-1(hi) ILC progenitor and defines its development pathway. Nature 539, 102–106 (2016).
Harly, C. et al. The transcription factor TCF-1 enforces commitment to the innate lymphoid cell lineage. Nat. Immunol. 20, 1150–1160 (2019).
Yagi, R. et al. The transcription factor GATA3 is critical for the development of All IL-7Ralpha-expressing innate lymphoid cells. Immunity 40, 378–388 (2014).
Aliahmad, P., de la Torre, B. & Kaye, J. Shared dependence on the DNA-binding factor TOX for the development of lymphoid tissue-inducer cell and NK cell lineages. Nat. Immunol. 11, 945–952 (2010).
Seehus, C. R. et al. The development of innate lymphoid cells requires TOX-dependent generation of a common innate lymphoid cell progenitor. Nat. Immunol. 16, 599–608 (2015).
Seillet, C. et al. Nfil3 is required for the development of all innate lymphoid cell subsets. J. Exp. Med. 211, 1733–1740 (2014).
Geiger, T. L. et al. Nfil3 is crucial for development of innate lymphoid cells and host protection against intestinal pathogens. J. Exp. Med. 211, 1723–1731 (2014).
Goh, W. & Huntington, N. D. Regulation of murine natural killer cell development. Front. Immunol. 8, 130 (2017).
Kwong, B. et al. T-bet-dependent NKp46+ innate lymphoid cells regulate the onset of TH17-induced neuroinflammation. Nat. Immunol. 18, 1117–1127 (2017).
Pearce, E. L. et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 302, 1041–1043 (2003).
Fuchs, A. et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-Producing Cells. Immunity 38, 769–781 (2013).
Townsend, M. J. et al. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity 20, 477–494 (2004).
Sojka, D. K. et al. Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. eLife 3, e01659 (2014).
Mackay, L. K. et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463 (2016).
Weizman, O. E. et al. ILC1 confer early host protection at initial sites of viral infection. Cell 171, 795–808 (2017).
Spooner, C. J. et al. Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat. Immunol. 14, 1229–1236 (2013).
Wong, S. H. et al. Transcription factor RORalpha is critical for nuocyte development. Nat. Immunol. 13, 229–236 (2012).
Walker, J. A. et al. Bcl11b is essential for group 2 innate lymphoid cell development. J. Exp. Med. 212, 875–882 (2015).
Ho, I. C., Tai, T. S. & Pai, S. Y. GATA3 and the T-cell lineage: essential functions before and after T-helper-2-cell differentiation. Nat. Rev. Immunol. 9, 125–135 (2009).
Cupedo, T. et al. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol. 10, 66–74 (2009).
Sanos, S. L. et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat. Immunol. 10, 83–91 (2009).
Zhong, C. et al. Group 3 innate lymphoid cells continuously require the transcription factor GATA-3 after commitment. Nat. Immunol. 17, 169–178 (2016).
Kiss, E. A. & Diefenbach, A. Role of the Aryl hydrocarbon receptor in controlling maintenance and functional programs of RORgammat(+) innate lymphoid cells and intraepithelial lymphocytes. Front. Immunol. 3, 124 (2012).
Lee, J. S. et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 13, 144–151 (2012).
Powell, N. et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor(+) innate lymphoid cells. Immunity 37, 674–684 (2012).
Parker, M. E. et al. c-Maf regulates the plasticity of group 3 innate lymphoid cells by restraining the type 1 program. J. Exp. Med. https://doi.org/10.1084/jem.20191030 (2019).
Tizian, C., et al. c-Maf restrains T-bet-driven programming of CCR6-negative group 3 innate lymphoid cells. eLife https://doi.org/10.7554/eLife.52549 (2020).
Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 449, 819–826 (2007).
Diefenbach, A. & Raulet, D. H. Strategies for target cell recognition by natural killer cells. Immunol. Rev. 181, 170–184 (2001).
Zafirova, B. et al. Altered NK cell development and enhanced NK cell-mediated resistance to mouse cytomegalovirus in NKG2D-Deficient Mice. Immunity 31, 270–282 (2009).
Kim, S. et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 436, 709–713 (2005).
Karre, K. Natural killer cell recognition of missing self. Nat. Immunol. 9, 477–480 (2008).
Hammer, Q. et al. Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat. Immunol. 19, 453–463 (2018).
Raulet, D. H. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 3, 781–790 (2003).
Brandt, C. S. et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 206, 1495–1503 (2009).
Moretta, A., Biassoni, R., Bottino, C., Mingari, M. C. & Moretta, L. Natural cytotoxicity receptors that trigger human NK-cell-mediated cytolysis. Immunol. Today 21, 228–234 (2000).
Barrow, A. D. et al. Natural killer cells control tumor growth by sensing a growth factor. Cell 172, 534–548 (2018).
Narni-Mancinelli, E., et al. Complement factor P is a ligand for the natural killer cell-activating receptor NKp46. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aam9628 (2017).
Kennedy, M. K. et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J. Exp. Med. 191, 771–780 (2000).
Lucas, M., Schachterle, W., Oberle, K., Aichele, P. & Diefenbach, A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 26, 503–517 (2007).
Kobayashi, M. et al. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J. Exp. Med. 170, 827–845 (1989).
Eisenring, M., vom Berg, J., Kristiansen, G., Saller, E. & Becher, B. IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat. Immunol. 11, 1030–1038 (2010).
Quatrini, L. et al. Host resistance to endotoxic shock requires the neuroendocrine regulation of group 1 innate lymphoid cells. J. Exp. Med. 214, 3531–3541 (2017).
Quatrini, L. et al. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat. Immunol. 19, 954–962 (2018).
Straub, T. et al. Bacterial coinfection restrains antiviral CD8 T-cell response via LPS-induced inhibitory NK cells. Nat. Commun. 9, 4117 (2018).
Waggoner, S. N., Cornberg, M., Selin, L. K. & Welsh, R. M. Natural killer cells act as rheostats modulating antiviral T cells. Nature 481, 394–398 (2011).
Biron, C. A., Byron, K. S. & Sullivan, J. L. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 320, 1731–1735 (1989).
Kagi, D. et al. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 369, 31–37 (1994).
Presti, R. M., Pollock, J. L., Dal Canto, A. J., O’Guin, A. K. & Virgin, H. Wt Interferon gamma regulates acute and latent murine cytomegalovirus infection and chronic disease of the great vessels. J. Exp. Med. 188, 577–588 (1998).
Krmpotic, A., Bubic, I., Polic, B., Lucin, P. & Jonjic, S. Pathogenesis of murine cytomegalovirus infection. Microbes Infect. 5, 1263–1277 (2003).
Diefenbach, A., Jamieson, A. M., Liu, S. D., Shastri, N. & Raulet, D. H. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat. Immunol. 1, 119–126 (2000).
Diefenbach, A., Jensen, E. R., Jamieson, A. M. & Raulet, D. H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 413, 165–171 (2001).
Guerra, N. et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 28, 571–580 (2008).
Waldhauer, I. et al. Tumor-associated MICA is shed by ADAM proteases. Cancer Res. 68, 6368–6376 (2008).
Deng, W. et al. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science 348, 136–139 (2015).
Sanchez-Correa, B. et al. Modulation of NK cells with checkpoint inhibitors in the context of cancer immunotherapy. Cancer Immunol. Immunother. 68, 861–870 (2019).
Takeda, K. et al. TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood 105, 2082–2089 (2005).
Vosshenrich, C. A. et al. A thymic pathway of mouse natural killer cell development characterized by expression of GATA-3 and CD127. Nat. Immunol. 7, 1217–1224 (2006).
O’Sullivan, T. E. et al. Adipose-resident group 1 innate lymphoid cells promote obesity-associated insulin resistance. Immunity 45, 428–441 (2016).
Doisne, J. M. et al. Composition, development, and function of uterine innate lymphoid cells. J. Immunol. 195, 3937–3945 (2015).
Almeida, F. F. et al. A point mutation in the Ncr1 signal peptide impairs the development of innate lymphoid cell subsets. Oncoimmunology 7, e1475875 (2018).
Turchinovich, G., Ganter, S., Barenwaldt, A. & Finke, D. NKp46 calibrates tumoricidal potential of type 1 innate lymphocytes by regulating TRAIL expression. J. Immunol. 200, 3762–3768 (2018).
Merino, D., Lalaoui, N., Morizot, A., Solary, E. & Micheau, O. TRAIL in cancer therapy: present and future challenges. Expert Opin. Ther. Targets 11, 1299–1314 (2007).
O’Leary, J. G., Goodarzi, M., Drayton, D. L. & von Andrian, U. H. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat. Immunol. 7, 507–516 (2006).
Wang, X. et al. Memory formation and long-term maintenance of IL-7Ralpha(+) ILC1s via a lymph node-liver axis. Nat. Commun. 9, 4854 (2018).
Paust, S. et al. Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat. Immunol. 11, 1127–1135 (2010).
Martinez-Gonzalez, I. et al. Allergen-experienced group 2 innate lymphoid cells acquire memory-like properties and enhance allergic lung inflammation. Immunity 45, 198–208 (2016).
Weizman, O. E. et al. Mouse cytomegalovirus-experienced ILC1s acquire a memory response dependent on the viral glycoprotein m12. Nat. Immunol. 20, 1004–1011 (2019).
Abt, M. C. et al. Innate immune defenses mediated by two ILC subsets are critical for protection against acute clostridium difficile infection. Cell Host Microbe. 18, 27–37 (2015).
Nabekura, T., Riggan, L., Hildreth, A. D., O’Sullivan, T. E. & Shibuya, A. Type 1 innate lymphoid cells protect mice from acute liver injury via interferon-gamma secretion for upregulating Bcl-xL expression in hepatocytes. Immunity 52, 96–108 (2020).
Denney, L. et al. Pulmonary epithelial cell-derived cytokine TGF-beta1 is a critical cofactor for enhanced innate lymphoid cell function. Immunity 43, 945–958 (2015).
Meylan, F. et al. The TNF-family cytokine TL1A promotes allergic immunopathology through group 2 innate lymphoid cells. Mucosal. Immunol. 7, 958–968 (2014).
Motomura, Y. et al. Basophil-derived interleukin-4 controls the function of natural helper cells, a member of ILC2s, in lung inflammation. Immunity 40, 758–771 (2014).
Gerbe, F. et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529, 226–230 (2016).
Howitt, M. R. et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351, 1329–1333 (2016).
Schneider, C. et al. A metabolite-triggered tuft Cell-ILC2 circuit drives small intestinal remodeling. Cell 174, 271–284 (2018).
von Moltke, J., Ji, M., Liang, H. E. & Locksley, R. M. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 529, 221–225 (2016).
Nadjsombati, M. S. et al. Detection of succinate by intestinal tuft cells triggers a Type 2 innate immune circuit. Immunity 49, 33–41 (2018).
Flamar, A. L., et al. Interleukin-33 induces the enzyme tryptophan hydroxylase 1 to promote inflammatory group 2 innate lymphoid cell-mediated immunity. Immunity https://doi.org/10.1016/j.immuni.2020.02.009 (2020).
Mahlakoiv, T., et al. Stromal cells maintain immune cell homeostasis in adipose tissue via production of interleukin-33. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aax0416 (2019).
Spallanzani, R. G., et al. Distinct immunocyte-promoting and adipocyte-generating stromal components coordinate adipose tissue immune and metabolic tenors. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aaw3658 (2019).
Rana, B. M. J. et al. A stromal cell niche sustains ILC2-mediated type-2 conditioning in adipose tissue. J. Exp. Med. 216, 1999–2009 (2019).
Dahlgren, M. W. et al. Adventitial stromal cells define group 2 innate lymphoid cell tissue niches. Immunity 50, 707–722 (2019).
Doherty, T. A. et al. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J. Allergy Clin. Immunol. 132, 205–213 (2013).
Tait Wojno, E. D. et al. The prostaglandin D2 receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal. Immunol. 8, 1313–1323 (2015).
Barnig, C. & Levy, B. D. Innate immunity is a key factor for the resolution of inflammation in asthma. Eur. Respiratory Rev. 24, 141–153 (2015).
Maric, J. et al. Prostaglandin E2 suppresses human group 2 innate lymphoid cell function. J. Allergy Clin. Immunol. 141, 1761–1773 (2018).
Duerr, C. U. et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat. Immunol. 17, 65–75 (2016).
Molofsky, A. B. et al. Interleukin-33 and interferon-gamma counter-regulate group 2 innate lymphoid cell activation during immune perturbation. Immunity 43, 161–174 (2015).
Laffont, S. et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J. Exp. Med. 214, 1581–1592 (2017).
Spencer, S. P. et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science 343, 432–437 (2014).
Kiss, E. A. et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334, 1561–1565 (2011).
van de Pavert, S. A. et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature 508, 123–127 (2014).
Li, S. et al. Aryl hydrocarbon receptor signaling cell intrinsically inhibits intestinal group 2 innate lymphoid cell function. Immunity 49, 915–928 (2018).
Grundemann, C. et al. The NK receptor KLRG1 is dispensable for virus-induced NK and CD8+ T-cell differentiation and function in vivo. Eur. J. Immunol. 40, 1303–1314 (2010).
Salimi, M. et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 210, 2939–2950 (2013).
Galle-Treger, L. et al. Costimulation of type-2 innate lymphoid cells by GITR promotes effector function and ameliorates type 2 diabetes. Nat. Commun. 10, 713 (2019).
Maazi, H. et al. ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity 42, 538–551 (2015).
Rauber, S. et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat. Med. 23, 938–944 (2017).
Wilhelm, C. et al. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat. Immunol. 12, 1071–1077 (2011).
Taylor, S. et al. PD-1 regulates KLRG1(+) group 2 innate lymphoid cells. J. Exp. Med. 214, 1663–1678 (2017).
Pelly, V. S. et al. IL-4-producing ILC2s are required for the differentiation of TH2 cells following Heligmosomoides polygyrus infection. Mucosal. Immunol. 9, 1407–1417 (2016).
Turner, J. E. et al. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J. Exp. Med. 210, 2951–2965 (2013).
Molofsky, A. B. et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J. Exp. Med. 210, 535–549 (2013).
Palm, N. W., Rosenstein, R. K. & Medzhitov, R. Allergic host defences. Nature 484, 465–472 (2012).
Yasuda, K. et al. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc. Natl. Acad. Sci. USA 109, 3451–3456 (2012).
Wilhelm, C. et al. Critical role of fatty acid metabolism in ILC2-mediated barrier protection during malnutrition and helminth infection. J. Exp. Med. 213, 1409–1418 (2016).
Harvie, M. et al. The lung is an important site for priming CD4 T-cell-mediated protective immunity against gastrointestinal helminth parasites. Infect. Immun. 78, 3753–3762 (2010).
Fallon, P. G. et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J. Exp. Med. 203, 1105–1116 (2006).
Oliphant, C. J. et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 41, 283–295 (2014).
Liu, T. et al. Type 2 innate lymphoid cells: A novel biomarker of eosinophilic airway inflammation in patients with mild to moderate asthma. Respiratory Med. 109, 1391–1396 (2015).
Kim, B. S. et al. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci. Transl. Med. 5, 170ra116 (2013).
Chang, Y. J. et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat. Immunol. 12, 631–638 (2011).
Halim, T. Y., Krauss, R. H., Sun, A. C. & Takei, F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 36, 451–463 (2012).
Halim, T. Y. et al. Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat. Immunol. 17, 57–64 (2016).
Halim, T. Y. et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40, 425–435 (2014).
Roediger, B. et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat. Immunol. 14, 564–573 (2013).
Camberis, M., Le Gros, G. & Urban, J. Jr. Animal model of nippostrongylus brasiliensis and heligmosomoides polygyrus. Curr. Protoc. Immunol. Chapter 19, Unit 19.12, John Wiley & Sons, Inc. (2003).
Monticelli, L. A. et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 12, 1045–1054 (2011).
Monticelli, L. A. et al. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl Acad. Sci. USA 112, 10762–10767 (2015).
Neumann, K. et al. A proinflammatory role of type 2 innate lymphoid cells in murine immune-mediated hepatitis. J. Immunol. 198, 128–137 (2017).
Rak, G. D. et al. IL-33-dependent group 2 innate lymphoid cells promote cutaneous wound healing. J. Invest. Dermatol. 136, 487–496 (2016).
McHedlidze, T. et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 39, 357–371 (2013).
van de Pavert, S. A. & Mebius, R. E. New insights into the development of lymphoid tissues. Nat. Rev. Immunol. 10, 664–674 (2010).
Ota, N. et al. IL-22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with Citrobacter rodentium. Nat. Immunol. 12, 941–948 (2011).
Kruglov, A. A. et al. Nonredundant function of soluble LTalpha3 produced by innate lymphoid cells in intestinal homeostasis. Science 342, 1243–1246 (2013).
Tsuji, M. et al. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity 29, 261–271 (2008).
Ibiza, S. et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature 535, 440–443 (2016).
Hepworth, M. R. et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science 348, 1031–1035 (2015).
Zhou, L. et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 568, 405–409 (2019).
Reynders, A. et al. Identity, regulation and in vivo function of gut NKp46+RORgammat+ and NKp46+RORgammat- lymphoid cells. EMBO J. 30, 2934–2947 (2011).
Longman, R. S. et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J. Exp. Med. 211, 1571–1583 (2014).
Castellanos, J. G. et al. Microbiota-induced TNF-like ligand 1A drives group 3 innate lymphoid cell-mediated barrier protection and intestinal T cell activation during colitis. Immunity 49, 1077–1089 (2018).
Hernandez, P. P. et al. Interferon-lambda and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat. Immunol. 16, 698–707 (2015).
Kim, M. H., Taparowsky, E. J. & Kim, C. H. Retinoic acid differentially regulates the migration of innate lymphoid cell subsets to the gut. Immunity 43, 107–119 (2015).
Satoh-Takayama, N. et al. The chemokine receptor CXCR6 controls the functional topography of interleukin-22 producing intestinal innate lymphoid cells. Immunity 41, 776–788 (2014).
Emgard, J. et al. Oxysterol sensing through the receptor GPR183 promotes the lymphoid-tissue-inducing function of innate lymphoid cells and colonic inflammation. Immunity 48, 120–132 (2018).
Chu, C. et al. Anti-microbial functions of group 3 Innate lymphoid cells in gut-associated lymphoid tissues are regulated by g-protein-coupled receptor 183. Cell Rep. 23, 3750–3758 (2018).
Chun, E. et al. Metabolite-sensing receptor ffar2 regulates colonic group 3 innate lymphoid cells and gut immunity. Immunity 51, 871–884 (2019).
Duffin, R. et al. Prostaglandin E(2) constrains systemic inflammation through an innate lymphoid cell-IL-22 axis. Science 351, 1333–1338 (2016).
Wolk, K. et al. IL-22 increases the innate immunity of tissues. Immunity 21, 241–254 (2004).
Goto, Y. et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science 345, 1254009 (2014).
Sonnenberg, G. F., Monticelli, L. A., Elloso, M. M., Fouser, L. A. & Artis, D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 34, 122–134 (2011).
Satpathy, A. T. et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat. Immunol. 14, 937–948 (2013).
Rankin, L. C. et al. Complementarity and redundancy of IL-22-producing innate lymphoid cells. Nat. Immunol. 17, 179–186 (2016).
Song, C. et al. Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. J. Exp. Med. 212, 1869–1882 (2015).
Sonnenberg, G. F., Fouser, L. A. & Artis, D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 12, 383–390 (2011).
Gronke, K. et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature 566, 249–253 (2019).
Hanash, A. M. et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity 37, 339–350 (2012).
Aparicio-Domingo, P. et al. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J. Exp. Med. 212, 1783–1791 (2015).
Huber, S. et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 491, 259–263 (2012).
Okada, S. et al. IMMUNODEFICIENCIES. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science 349, 606–613 (2015).
Lim, A. I. et al. Systemic human ILC precursors provide a substrate for tissue ILC differentiation. Cell 168, 1086–1100 (2017).
Gladiator, A., Wangler, N., Trautwein-Weidner, K. & LeibundGut-Landmann, S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J. Immunol. 190, 521–525 (2013).
Kim, H. Y. et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat. Med. 20, 54–61 (2014).
Seo, G. Y. et al. LIGHT-HVEM signaling in innate lymphoid cell subsets protects against enteric bacterial infection. Cell Host microbe 24, 249–260 e244 (2018).
Buonocore, S. et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464, 1371–1375 (2010).
Mortha, A. et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 343, 1249288 (2014).
Magri, G. et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat. Immunol. 15, 354–364 (2014).
Hepworth, M. R. et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 498, 113–117 (2013).
von Burg, N. et al. Activated group 3 innate lymphoid cells promote T-cell-mediated immune responses. Proc. Natl. Acad. Sci. USA 111, 12835–12840 (2014).
Melo-Gonzalez, F. et al. Antigen-presenting ILC3 regulate T cell-dependent IgA responses to colonic mucosal bacteria. J. Exp. Med. 216, 728–742 (2019).
Schwartz, C. et al. ILC2s regulate adaptive Th2 cell functions via PD-L1 checkpoint control. J. Exp. Med. 214, 2507–2521 (2017).
Klose, C. S. & Artis, D. Neuronal regulation of innate lymphoid cells. Curr. Opin. Immunol. 56, 94–99 (2018).
Veiga-Fernandes, H. & Mucida, D. Neuro-Immune Interactions at Barrier Surfaces. Cell 165, 801–811 (2016).
Oetjen, L. K. et al. Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell 171, 217–228 (2017).
Wilson, S. R. et al. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 155, 285–295 (2013).
Nagashima, H. et al. Neuropeptide CGRP limits group 2 innate lymphoid cell responses and constrains type 2 inflammation. Immunity 51, 682–695 (2019).
Wallrapp, A. et al. Calcitonin gene-related peptide negatively regulates alarmin-driven type 2 innate lymphoid cell responses. Immunity 51, 709–723 (2019).
Xu, H. et al. Transcriptional atlas of intestinal immune cells reveals that neuropeptide alpha-CGRP modulates group 2 innate lymphoid cell responses. Immunity 51, 696–708 (2019).
Sui, P., et al. Pulmonary neuroendocrine cells amplify allergic asthma responses. Science https://doi.org/10.1126/science.aan8546 (2018).
Galle-Treger, L. et al. Nicotinic acetylcholine receptor agonist attenuates ILC2-dependent airway hyperreactivity. Nat. Commun. 7, 13202 (2016).
Moriyama, S. et al. beta2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science 359, 1056–1061 (2018).
Dalli, J., Colas, R. A., Arnardottir, H. & Serhan, C. N. Vagal regulation of group 3 innate lymphoid cells and the immunoresolvent PCTR1 controls infection resolution. Immunity 46, 92–105 (2017).
Nussbaum, J. C. et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502, 245–248 (2013).
Talbot, S. et al. Silencing nociceptor neurons reduces allergic airway inflammation. Neuron 87, 341–354 (2015).
Klose, C. S. N. et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 549, 282–286 (2017).
Cardoso, V. et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 549, 277–281 (2017).
Wallrapp, A. et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549, 351–356 (2017).
Talbot, J., et al. Feeding-dependent VIP neuron-ILC3 circuit regulates the intestinal barrier. Nature https://doi.org/10.1038/s41586-020-2039-9 (2020).
Seillet, C., et al. The neuropeptide VIP confers anticipatory mucosal immunity by regulating ILC3 activity. Nat. Immunol. https://doi.org/10.1038/s41590-019-0567-y (2019).
Godinho-Silva, C. et al. Light-entrained and brain-tuned circadian circuits regulate ILC3s and gut homeostasis. Nature 574, 254–258 (2019).
Teng, F., et al. A circadian clock is essential for homeostasis of group 3 innate lymphoid cells in the gut. Sci Immunol. 4, (2019).
Wang, Q., et al. Circadian rhythm-dependent and circadian rhythm-independent impacts of the molecular clock on type 3 innate lymphoid cells. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aay7501 (2019).
Giacomin, P. R. et al. Epithelial-intrinsic IKKalpha expression regulates group 3 innate lymphoid cell responses and antibacterial immunity. J. Exp. Med. 212, 1513–1528 (2015).
Rafei-Shamsabadi, D. A. et al. Lack of type 2 innate lymphoid cells promotes a type I-driven enhanced immune response in contact hypersensitivity. J. Invest Dermatol. 138, 1962–1972 (2018).
Cella, M. et al. Subsets of ILC3-ILC1-like cells generate a diversity spectrum of innate lymphoid cells in human mucosal tissues. Nat. Immunol. 20, 980–991 (2019).
Bernink, J. H. et al. Interleukin-12 and -23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity 43, 146–160 (2015).
Gao, Y. et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 18, 1004–1015 (2017).
Cortez, V. S. et al. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-beta signaling. Nat. Immunol. 18, 995–1003 (2017).
Park, E., et al. Toxoplasma gondii infection drives conversion of NK cells into ILC1-like cells. eLife https://doi.org/10.7554/eLife.47605 (2019).
Bal, S. M. et al. IL-1beta, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat. Immunol. 17, 636–645 (2016).
Golebski, K. et al. IL-1beta, IL-23, and TGF-beta drive plasticity of human ILC2s towards IL-17-producing ILCs in nasal inflammation. Nat. Commun. 10, 2162 (2019).
Zhang, K. et al. Cutting edge: notch signaling promotes the plasticity of group-2 innate lymphoid cells. J. Immunol. 198, 1798–1803 (2017).
Bernink, J. H. et al. c-Kit-positive ILC2s exhibit an ILC3-like signature that may contribute to IL-17-mediated pathologies. Nat. Immunol. 20, 992–1003 (2019).
Seehus, C. R. et al. Alternative activation generates IL-10 producing type 2 innate lymphoid cells. Nat. Commun. 8, 1900 (2017).
Bando, J. K. et al. ILC2s are the predominant source of intestinal ILC-derived IL-10. J. Exp. Med. 217, e20191520 https://doi.org/10.1084/jem.20191520 (2020).
Miyamoto, C. et al. Runx/Cbfbeta complexes protect group 2 innate lymphoid cells from exhausted-like hyporesponsiveness during allergic airway inflammation. Nat. Commun. 10, 447 (2019).
Wang, S. et al. Regulatory innate lymphoid cells control innate intestinal inflammation. Cell 171, 201–216 (2017).
Brestoff, J. R. & Artis, D. Immune regulation of metabolic homeostasis in health and disease. Cell 161, 146–160 (2015).
Brestoff, J. R. et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 519, 242–246 (2015).
Lee, M. W. et al. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell 160, 74–87 (2015).
Odegaard, J. I. et al. Perinatal licensing of thermogenesis by IL-33 and ST2. Cell 166, 841–854 (2016).
Dalmas, E. et al. Interleukin-33-activated islet-resident innate lymphoid cells promote insulin secretion through myeloid cell retinoic acid production. Immunity 47, 928–942 (2017).
Wang, X. et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature 514, 237–241 (2014).
Vely, F. et al. Evidence of innate lymphoid cell redundancy in humans. Nat. Immunol. 17, 1291–1299 (2016).
Acknowledgements
We thank Artis Lab members for thoughtful discussions and Drs. Anne-Laure Flamar, Mohammad Arifuzzaman and Claudia Dürr for critically reading the manuscript. The work was supported by grants from the German Research Foundation (DFG; KL 2963/2-1 and SPP1937 - KL 2963/3-1 to C.S.N.K.) and the European Research Council (ERCEA; ERC Starting Grant #803087 to C.S.N.K.), the National Institutes of Health (NIH; AI074878, AI095466, AI095608 and AI102942 to D.A.), the Burroughs Wellcome Fund (to D.A.) and the Crohn’s & Colitis Foundation of America (to D.A.), Cure for IBD (to D.A.) and the Rosanne H. Silberman Foundation (to D.A.).
Author information
Authors and Affiliations
Contributions
C.S.N.K. and D.A. wrote the manuscript together.
Corresponding authors
Ethics declarations
Competing interests
D.A. has contributed to scientific advisory boards at Pfizer, Takeda, FARE, and the KRF in the last twelve months. C.S.N.K. has no competing interests to declare.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Klose, C.S.N., Artis, D. Innate lymphoid cells control signaling circuits to regulate tissue-specific immunity. Cell Res 30, 475–491 (2020). https://doi.org/10.1038/s41422-020-0323-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41422-020-0323-8
This article is cited by
-
Cooperation of ILC2s and TH2 cells in the expulsion of intestinal helminth parasites
Nature Reviews Immunology (2024)
-
The immunoregulatory roles of non-haematopoietic cells in the kidney
Nature Reviews Nephrology (2024)
-
Gegen Qinlian Decoction treatment of asymptomatic hyperuricemia by targeting circadian immune function
Chinese Medicine (2023)
-
Interleukin-9 production by type 2 innate lymphoid cells induces Paneth cell metaplasia and small intestinal remodeling
Nature Communications (2023)
-
mTORC2 acts as a gatekeeper for mTORC1 deficiency-mediated impairments in ILC3 development
Acta Pharmacologica Sinica (2023)
