
Abstract
Essential hypertension (EH) is a multifactorial disease of the cardiovascular system that is influenced by the interplay of genetic, epigenetic, and environmental factors. The molecular dynamics underlying EH etiopathogenesis is unknown; however, earlier studies have revealed EH-associated genetic variants. Nevertheless, this finding alone is not sufficient to explain the variability in blood pressure, suggesting that other risk factors are involved, such as epigenetic modifications. Therefore, this review highlights the potential contribution of well-defined epigenetic mechanisms in EH, specifically, DNA methylation, post-translational histone modifications, and microRNAs. We further emphasize global and gene-specific DNA methylation as one of the most well-studied hallmarks among all epigenetic modifications in EH. In addition, post-translational histone modifications, such as methylation, acetylation, and phosphorylation, are described as important epigenetic markers associated with EH. Finally, we discuss microRNAs that affect blood pressure by regulating master genes such as those implicated in the renin-angiotensin-aldosterone system. These epigenetic modifications, which appear to contribute to various cardiovascular diseases, including EH, may be a promising research area for the development of novel future strategies for EH prevention and therapeutics.
Similar content being viewed by others
Introduction
Almost 95% of adult patients with high blood pressure (BP) are believed to have essential or idiopathic hypertension. Moreover, essential hypertension (EH) is a target of considerable public health concern, as EH affects over one billion people globally [1]. High BP and associated healthcare and management costs in the United States alone amount to $46 billion annually [2]. Despite such a high healthcare expenditure, in 2013, over 360,000 American deaths were due to EH as a prime contributing cause [2], accounting for almost a thousand deaths daily. Nonetheless, the etiopathogenesis of EH remains a mystery, making advancements in related therapies difficult. Therefore, elucidating the underlying mechanism(s) of EH is clinically essential for the development of novel therapeutic strategies [3, 4]. It is known that EH is affected by genetic, epigenetic, and environmental factors. Differential etiologies of EH are the product of dynamic interactions among genetic and environmental elements, leading to alteration of biological pathways and, ultimately, hypertension [5,6,7]. EH is one of the major risk factors for cardiovascular (CV) pathologic remodeling leading to heart failure or stroke [2, 8], renal injuries [9], and brain dysfunction [10].
Previously, limited association was thought to be present between genetic factors and hypertension [11]. Currently, numerous studies have demonstrated the significant effect of genetic factors on changes in BP and related pathologies [5, 8, 12]. Moreover, genome-wide association studies (GWAS) have identified common genetic variants in common population-based disease traits, such as EH [13,14,15,16]. Recently, a large genetic association study, consisting of one million subjects, identified 535 novel loci related to BP traits [17]. Nevertheless, single-gene, target-based treatment for EH remains unavailable to clinicians due to the polygenic behavior of EH [5,6,7]. Thus, it is evident that a conventional Mendelian genetic approach does not suffice to explain the regulation of BP [18, 19].
Studies suggested that epigenetics may play a crucial role in BP regulation [20]. For example, Kato et al. reported that EH can arise from epigenetic dysfunction [21]. Epigenetics, i.e., the way in which genes and the environment interact, can regulate gene expression to control a related phenotype (Fig. 1). Epigenetic changes are measurable [22] and can be substantially modulated at any life stage through environmental stimuli, such as drugs, nutrition, and age [23]. Thus, an environmentally predisposed epigenetic map [24] could help elucidate the unsolved questions that recent genetic approaches could not explain. Interestingly, reports have shown that conditions influenced by epigenetics, such as EH, can be reversed simply by making different lifestyle choices [25, 26], suggesting an array of treatment options for related disorders that are not always feasible for genetic diseases. For this reason, the global scientific community is investigating epigenetics as a key player in numerous pathophysiologies, including EH [19]. This notion is clearly reflected by worldwide initiatives such as the Human Epigenome Project [27], International Human Epigenome Consortium [28], and other related programs [13,14,15].

Epigenetic and genetic dynamics regulate individual phenotypes. In addition to heritable Mendelian genetics, polygenic phenotypes, such as essential hypertension, are significantly affected by gene-environment interactions triggering epigenetic modifications
It is anticipated that this review will serve as a comprehensive resource of the current literature on major epigenetic modifications during EH, highlighting the potential contribution of DNA methylation, post-translational histone modifications, and microRNAs (miRNAs) (Fig. 2; Table 1). Here, in addition to those associated with EH, we also present the epigenetic changes associated with pre-eclampsia, emphasizing the involvement of epigenetics in EH, which is comparable to high BP disorder.

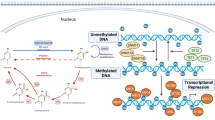
Epigenetic modifications in a mammalian system. a DNA methylation originates at specific genomic regions via transfer of a methyl group from S-adenosyl-L-methionine to the 5’-position of a cytosine ring to form 5-methylcytosine (5mCy). A chain of methylated cytosine and guanine dinucleotides form a CpG island that is often located in gene regulatory regions, such as a promoter, and is often negatively correlated with gene transcription. b Post-translational DNA modifications, such as histone tail acetylation in nucleosomes, support gene transcription via transforming condensed chromatin into the more lightly packed state of euchromatin. In contrast, histone tail deacetylation promotes the condensed state of heterochromatin to promote gene silencing. Similarly, less frequent histone modifications, such as methylation and phosphorylation, are observed, and these also affect gene transcription to some extent. c Another important mode of epigenetic modification is the small noncoding RNAs, such as microRNAs that negatively regulate the corresponding gene activation via inhibition of mRNA transcription and translation
EH and DNA methylation-mediated epigenetic regulation
DNA methylation is one of the most well-known epigenetic mechanisms of gene regulation in mammals [29], whereby a methyl group binds to the 5’ region of a DNA cytosine via a covalent bond [30]. The frequent short sequence of genomic DNA containing a series of cytosine and guanine dinucleotides connected via phosphodiester bonds (linear 5′-CpG-3′ sequence) is known as a ‘CpG island’ [31]. Functionally, hypermethylation of cytosine at such CpG sites, particularly in the gene promoter region, often leads to gene silencing. Such epigenetic markers also play a crucial role in defining the genetically active or inactive regions of the genome by regulating the interaction of DNA and transcription factors [32, 33]. However, not all mammalian cell types show the same degree of methylation, which can result in the expression of diverse phenotypes. Accordingly, Raftopoulos et al. reported the close association of the onset and severity of hypertension with the degree of genomic DNA methylation [23], suggesting the importance of future studies related to such epigenetic markers during EH.
Here, it is noteworthy that clinical studies related to EH and end-organ damage are restricted by the limited availability of relevant human samples. Thus, in a large cohort human study, it is generally impossible to access EH-affected tissue samples from subjects at risk, making peripheral blood the material of choice. However, disease progression and severity may also affect the metabolic pathways governing DNA methylation, leading to altered methylation patterns in different tissues [34]. Kato et al. reported on blood and cross-tissue patterns of DNA methylation and suggested that blood and other tissues (liver, muscle, subcutaneous and visceral fat) exhibit comparable methylation patterns [21]. These pioneering results suggest that DNA methylation markers observed in peripheral blood mononuclear cells (PBMCs) can provide a basis for proxy methylation patterns in other related tissues. Contrary to this notion, rat studies showed that different body tissues may acquire dissimilar methylation patterns in response to a specific drug exposure [35]. Therefore, the mere analysis of PBMCs, as epigenetic determinants, may not be helpful for studying other body tissues.
Epigenetic DNA methylation
DNA methylation can be categorized as global or gene-specific, depending on whether whole-genome 5-methylcytosine (5mCy) content is considered or methylation of a gene in a specific region [36]. The key function of DNA methylation, as an epigenetic marker, is to preserve genomic stability at the global level and regulation of mRNA expression at the gene level [37]. Several reports show the impact of DNA methylation in different pathologies, including EH, thus directing researchers’ attention toward DNA methylation in hypertension and related CV diseases [38, 39]. A summary of the reported epigenetic changes, including DNA methylation during EH, is presented in Table 1. In the following sections, we will highlight the two well-known forms of DNA methylation, such as cytosine methylation and cytosine hydroxymethylation, which can influence the genome at either the global or gene-specific level.
EH and Cytosine methylation
5mCy, accounting ~3–4% of all cytosine, are uniformly distributed throughout the genome of all mammalian cells [40]. Interestingly, studies have demonstrated a correlation between different pathological conditions and the differential expression of global and gene-specific 5mCy levels [37, 41]. Kato et al. found that 28 of 35 single nucleotide polymorphism variants identified through GWAS for hypertension are also associated with methylation markers [21], strongly suggesting the role of DNA methylation in EH.
EH and Cytosine Hydroxymethylation
Like 5mCy, 5-hydroxymethylcytosine (5hmCy), a byproduct of the demethylation process, is another major epigenetic marker in mammalian genomic DNA. Similar to 5mCy, 5hmCy is also distributed throughout the whole mammalian genome, with different levels in dissimilar tissues [40, 42]. Furthermore, several reports confirm that 5hmCy can contribute to gene regulation [43,44,45]. In addition, as initial evidence, a rat study showed a positive relationship between 5hmCy levels and hypertension [46]. However, currently, only limited data are available that tentatively associate EH and DNA hydroxymethylation. Therefore, future studies are needed to elucidate the role of 5hmCy in human EH.
EH and Global DNA methylation
A global 5mCy map defining the DNA methylation pattern is unique and can provide a blueprint for DNA behavior and its stability. Pioneering DNA methylation studies have confirmed the relationship between global 5mCy and EH [47]. Accordingly, Smolarek et al. showed that the levels of 5mCy in genomic DNA in peripheral blood are lower in hypertensive patients [47], suggesting a negative relationship between global DNA methylation levels and the severity of EH. In contrast, hypertension during pregnancy (pre-eclampsia) is reported to be positively associated with global DNA hypermethylation [48]. Such evidence clearly suggests that global DNA methylation may influence the progression of EH. In a study of young males, Wang et al. reported that global DNA methylation may play an important role in the pathogenesis of EH in an age-dependent manner [49]. A recent rat study showed that pressure overload-induced cardiac hypertrophy, a hallmark of target organ damage related to EH [50], can be significantly restored by inhibiting global DNA methylation [51], suggesting that DNA methylation plays a role during EH-associated CV damage.
EH and Gene-specific DNA methylation
Current research has mostly focused on the methylation pattern of specific gene regions. Gene-specific epigenetic changes arise in precise genomic regions, namely, CpG islands, frequently located in the gene promoter region [19]. CpG islands are found at the promoter of 40% of genes, while the remaining genomic regions are deprived of CpG sites [52]. Interestingly, in normal somatic cells, 90% of CpG sites are methylated; however, CpG sites at the promoter region are comparatively safer from such modifications [52]. Hypermethylated CpG islands, often in promoter regions, are responsible for gene transcription repression [23]. This notion is evident from various animal and human studies that demonstrate a negative correlation between 5mCy content and gene expression [53,54,55]; however, such a correlation is not always present and is still debatable [41]. Earlier reports revealed a positive association between 5hmCy levels and gene expression [46, 53,54,55,56], suggesting that lower 5mCy levels lead to gene silencing, whereas higher 5hmCy levels are associated with gene activation (Fig. 3). In addition, DNA methylation at a specific gene locus may modulate the interaction between gene transcription factors, or related epigenetic markers in histones, in a manner that leads to differential expression of the affected gene [57, 58].

Models of gene regulation via gene-specific epigenetic markers, such as 5-methylcytosine (5mCy) and 5-hydroxymethylcytosine (5hmCy). a series of methylated cytosine and guanine dinucleotides (CpG islands), often located in the gene regulatory promoter region, are inversely associated with gene transcription, causing gene silencing. In contrast, the promoter presence of multiple hydroxymethylated cytosine and guanine dinucleotides results in gene induction
Examples of gene-specific DNA methylation in EH
Several genes from the published literature have demonstrated that epigenetic changes are able to regulate pathways and biological processes identified during EH. These include sympathetic stimulation and renin-angiotensin-aldosterone system (RAAS) activation, leading to altered renal sodium reabsorption and vasoconstriction (Fig. 4). In fact, several RAAS genes are routinely targeted to study EH. Moreover, entire RAAS components exist in the brain and may show impaired function in many pathophysiological conditions, including EH. For instance, 11-Beta-hydroxysteroid dehydrogenase Type-2 (HSD11B2) catalyzes the conversion of cortisol to cortisone. Similar to aldosterone, cortisol regulates renal sodium reabsorption and thus plays a major role in the regulation of arterial BP [23]. Abnormal regulation of aldosterone and cortisol production can lead to EH, as seen in primary aldosteronism [59, 60]. HSD11B2 gene repression, due to promoter hypermethylation, leads to abnormal levels of cortisol- and cortisone-active metabolites (tetrahydrocortisol, tetrahydrocortisone), resulting in the onset of EH [39, 61]. Similarly, hypertensive adult rat model studies demonstrate a positive association between HSD11B2 gene promoter hypermethylation and hypertension [62]. In human newborns, the HSD11B2 gene promoter is hypermethylated, along with downregulation of HSD11B2 gene expression [63], suggesting the risk of EH [61] via abnormal renal sodium reabsorption. Recently, Takeda et al. showed that DNA methylation patterns present at gene promoters in adult somatic cells can be modulated to activate expression of the adrenal aldosterone synthase gene, Cytochrome P450 Family 11 Subfamily B Member 2 (CYP11B2) [64], which has an essential role in BP regulation [59]. In another study, Lee et al. emphasized the role of membrane transporter genes in hypertension pathogenesis, since alterations in ion flux mechanisms can influence BP regulation [65]. In spontaneously hypertensive rats (SHRs), this study also demonstrates that hypomethylation of the Na + /K + /2Cl- cotransporter 1 (NKCC1) gene promoter is associated with NKCC1 upregulation 65 correlated with postnatal hypertension [39, 66]. These studies indicate that changes due to the dynamic nature of DNA methylation can affect associated gene expression to regulate BP.

Epigenetic regulation of the RAAS pathway to modulate BP. a RAAS is one of the major regulators of BP, and its positive stimulation is associated with essential hypertension. In response to a low blood volume, liver-produced angiotensinogen is converted to angiotensin I by renal renin, which is further converted to active angiotensin II peptide via pulmonary ACE. Angiotensin II moves to different target organs via systemic flow and binds to its receptor AGTR1 to modulate different pathways, such as secretion of aldosterone (via CYP11B activation) and ADH from the adrenal and pituitary glands, respectively, and eNOS uncoupling, causing reduced NO production in arteries. Aldosterone causes the kidney to reabsorb sodium and water from urine and excrete potassium, resulting in increased BP, while high systemic ADH and low arterial NO act as vasoconstrictors to elevate BP. Here, plus (green) and minus (red) signs represent positive and negative regulation, respectively. b Diverse epigenetic determinants can influence and participate in the regulation of BP by activating, or silencing, the master genes associated with RAAS. As shown here, nearly all key RAAS genes are governed by one or multiple epigenetic mechanisms, suggesting the importance of epigenetic regulation in hypertension and the pathogenesis of related diseases. RAAS renin-angiotensin-aldosterone system, BP blood pressure, REN renin, AGT angiotensinogen, AGTR1 angiotensin II type 1 receptor, ACE angiotensin-converting enzyme, CYP11B cytochrome P450 Family 11 Subfamily B, ADH antidiuretic hormone, eNOS endothelial nitric oxide synthase, NO nitric oxide
Moreover, angiotensin I receptor antagonists effectively reduce BP in angiotensin II-dependent hypertension [67]. An in vitro study using human cells [NCI H295R adrenocortical cells] illustrates that interleukin-6 (IL-6)-induced hypomethylation of transcription start sites and enhancer-binding sites leads to activation of angiotensinogen (AGT) gene expression [68]. Aldosterone hormone regulates Na+ transport in renal collecting ducts to control water uptake, blood volume and, therefore, BP. Aldosterone overexpression leads to EH and CVD [60, 69]. Moreover, in response to a high-salt diet, hypomethylation of the AGT gene promoter supports AGT upregulation in rat visceral adipose tissue, leading to hypertension [68]. In another report, rat studies of a maternal low protein diet show that elevated expression of the angiotensin receptor AT1B gene in adrenal glands, due to hypomethylation of the AT1B gene, promotes salt-responsive hypertension [70]. Moreover, as a master regulator of BP, angiotensin-converting enzyme (ACE) converts angiotensin I to active angiotensin II [35], a peptide with potent vasoconstrictive properties that cause blood vessels to narrow, leading to higher BP. A study by Fan et al. showed that aberrant methylation of the ACE2 gene promoter may be associated with EH [71]. Interestingly, studies from rat (lungs and liver) and in vitro models [such as human liver (HepG2), colon (HT29), microvascular endothelial (HMEC-1), and lung (SUT) cell lines] demonstrate that hypermethylation of the ACE gene promoter results in its transcriptional repression, indicating the epigenetic regulation of ACE-mediated hypertension [35]. Similarly, during pregnancy, maternal protein deficiency and concurrent hypomethylation of ACE result in fetal hypertension and cognitive dysfunction [72]. Litwin et al. demonstrated elevated mRNA expression of ACE and repression of AGT in young individuals with hypertension [73]. Additional human studies show that EH is associated with hypomethylation of the angiotensin II type 1 receptor (AGTR1) gene [74, 75]. Additionally, studies conducted in SHRs reveal that hypertension during aging is related to upregulation of angiotensin 1a receptor (Atgr1a) gene expression in endothelial cells induced by hypomethylation of the Atgr1a gene promoter [76]. Angiotensin II also induces the secretion of antidiuretic hormone (ADH) from the pituitary gland, which is well known for osmoregulation and acts as a vasoconstrictor to increase BP [77]. Reports show that the DNA methylation pattern of specific CpG sites within the ADH gene promoter can affect ADH mRNA expression in rat brains [78, 79]. Collectively, this evidence suggests that ADH dysregulation from epigenetic modifications may affect BP and, as suggested by other studies [80, 81], may also play a role in the onset of moderate hypertension. Since stimulation of the sympathetic nervous system (SNS) leads to alterations in the arterial BP-regulating RAAS pathway [70], it is not surprising that alterations in the methylation of associated genes, such as AGT, renin (REN), ACE, and HSD11B2, are related to BP levels. Similarly, alterations in the RAAS pathway lead to changes in sodium reabsorption in the kidney, hypermethylation of sodium channel epithelial 1 alpha subunit (SCNN1A) [82], and differential methylation of sodium channel epithelial 1 beta subunit (SCNN1B) [83] in gene promoters, all of which have been reported to be associated with EH. Therefore, it is not surprising that genetic variants and altered epigenetic regulation in the RAAS pathway have been well established as having regulatory control over EH. In addition, other studies have suggested that scavenging of reactive oxygen species (ROS) can restore endothelial function in animal models of hypertension and atherosclerosis [84]. Endothelial nitric oxide synthase (eNOS) plays a key role in arterial BP regulation by modulating vascular tone through the production of nitric oxide (NO) in the vascular endothelium [85, 86]. Endothelial dysfunction characterized by reduced NO production is a hallmark of hypertension [86]. Studies reveal that eNOS uncoupling (transforming eNOS into a pro-oxidant enzyme) in angiotensin II-treated mice significantly reduces aortic NO synthesis, resulting in elevated BP [87]. In vitro studies reveal that hypomethylation of the eNOS gene promoter is responsible for activation of eNOS expression in human endothelial cells [88], suggesting that epigenetic regulation of eNOS contributes to human hypertension. In addition, epigenetically regulated angiotensin II [35] activates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which causes ROS generation that may, in turn, result in endothelial dysfunction [89] and, therefore, may be associated with abnormal BP. Interestingly, other studies reported that the major source of ROS in the right atrial appendage of atrial fibrillation results from a rise in myocardial NADPH oxidase activity, rather than eNOS uncoupling [90].
Aside from the abovementioned genes in the RAAS pathway, several other documented genes are associated with EH. For example, reports show that gestational hypoxia induces promoter hypermethylation and resultant estrogen receptor-α (ERα) gene silencing in sheep uterine arteries, favoring an increased risk of pre-eclampsia [91]. Similarly, offspring from vitamin D-deficient Sprague–Dawley (SD) rats show signs of EH correlated with hypermethylation of the Pannexin 1 (PANX1) gene promoter [92]. Genome-wide methylation studies conducted in young African males using blood leukocytes reveal that the sulfatase 1 (SULF1) gene is hypermethylated to regulate chromatin activation/deactivation in EH patients [49]. Methyl CpG binding protein 2 (MECP-2)-mediated hypermethylation of the norepinephrine transporter (NET) gene promoter leads to its repression, causing a blockage in norepinephrine, a BP-regulating systemic catecholamine [93], and dopamine transport to the presynaptic neuron, leading to EH [94]. Moreover, during the early stage of pre-eclampsia, hypomethylation of the promoter region of the tissue inhibitor of metalloproteinases 3 (TIMP3) gene has been documented [95]. Similarly, hypomethylation of the Serine Protease Inhibitor A3 (SERPINA3) [96] and Cullin-7 (CUL7) [97] gene promoters is associated with hypertension during pregnancy. Hypomethylation of the α-adducin (ADD1) gene promoter sequence was reported to be associated with EH risk [98, 99]. Recently, Jin et al. showed that mitofusin 2 (Mfn2), a gene that inhibits vascular smooth muscle cell proliferation and is involved in BP regulation, is downregulated in hypertensive patients with a concurrent lower level of DNA methylation of the full-length Mfn2 gene [100]. Other similar studies in hypertensive patients suggest that hypomethylation of genes such as proinflammatory cytokine IL-6 [101], immune receptor toll‑like receptors (TLRs) [102], and systemic inflammatory response mediator interferon-γ (IFN-γ) [103] increases the risk of EH. These studies also demonstrate an interesting aspect of epigenetic regulation in that a CpG island could have multiple differentially methylated CpG sites, from which a certain cytosine methylation level could determine the onset of EH [99,100,101,102] in an age-, sex- and therapy-specific manner [83, 99, 101]. Similarly, Fan et al. reported that differential methylation levels of the glucose metabolism regulator glucokinase (GCK) gene are associated with the onset of EH [104], suggesting that diabetes is a potential risk factor for EH progression.
In summary, gene-specific DNA methylation and the associated diverse range of biological pathways may play a significant role in EH. For that reason, the epigenetic markers could be used to estimate the levels and the associated risk of EH and, ultimately, its pathogenesis.
EH and Histone modification-mediated epigenetic regulation
Post-translational modifications at genomic DNA N-terminal histone tail sites (Fig. 2), such as methylation, acetylation, phosphorylation, and ubiquitination, are important epigenetic regulators related to hypertension [105,106,107,108]. Specific histone modification has a distinctive effect on the corresponding chromatin dynamics, often with comparable outcomes. For example, histone acetylation mostly activates gene transcription, while histone deacetylation favors gene silencing. However, such a correlation is not always true and is still controversial.
Histone lysine methylation at location 79 (H3K79) represses gene transcription, while histone arginine methylation enhances it. Hypermethylation of the histone lysine at location 9 (K9) leads to gene repression, while its hypomethylation activates gene transcription [31, 109]. In these modifications, interaction dynamics between histone tails and epigenetic factors control the degree of DNA wrapping around histones to regulate the transcription of the associated genes. These dynamics can also create an interactive site for chromatin-altering enzymes to activate specific gene expression [110]. In addition, previous reports reveal that epigenetic markers could be transgenerationally inherited [111]. Accordingly, a recent report suggests the paternal mode of histone-based epigenetic inheritance that determines the fertility of the future progeny [112], thus providing a potential mechanism to show how the paternal epigenetic map may affect the development and health of the offspring.
Examples of epigenetic Histone modifications in EH
Previous studies of hypertensive rats reveal that histone modifications are associated with ACE1 upregulation [113]. Similarly, hypertensive offspring from lipopolysaccharide-treated rats show ACE1 gene overexpression associated with histone H3 acetylation (H3Ac) in the promoter region of ACE1 [114]. Another related study using hypertensive adult rats demonstrated downregulation of the HSD11B2 gene associated with a decrease in H3K36 trimethylation (H3K36me3), suggesting a role of histone modification-mediated modulation of chromatin structure during hypertension [62]. In vitro studies using human umbilical vein endothelial cells (HUVECs) reveal that hyperacetylation of H3K9 and H4K12 and di- and trimethylation of H3K4 at the eNOS gene promoter result in upregulation of BP-regulating eNOS gene expression [85, 86, 115]. This result suggests that changes in eNOS mRNA levels in response to histone acetylation may play a critical role in human EH. Cho et al. reported that angiotensin II-triggered upregulation of NKCC1 mRNA and protein in rat aortas is correlated with histone H3 and hypomethylation [116], suggesting that epigenetic modifications regulate NKCC1 transcription and associated sodium renal reabsorption to affect BP. Disruptors of telomeric silencing (DOT) are associated with hypertension [105]. In response to hydrochlorothiazide, patients have shown a strong relationship between DOT1-like histone H3K79 methyltransferase (DOT1L) and hypertension [117]. Similarly, DOT1a interaction with Af9 (leukemia gene located at chromosome 9) induces H3K79 hypermethylation, causing repression of the renal epithelial sodium channel (ENaC-a) gene, leading to maintenance of normal/lower BP. However, aldosterone-mediated disruption of DOT1a-Af9 interaction leads to hypomethylation of H3K79, resulting in ENaC-a gene activation and, finally, high BP [118]. Mehrotra et al. showed that EH-mediated end-organ damage, such as cardiac hypertrophy in salt-induced hypertensive rats, is mediated by higher levels of AcH4 and H3K4me3, but lower levels of H3K27me3 and H3K9me3, in overexpressed atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) gene promoters [119].
Moreover, in addition to the kidneys, the central nervous system (CNS) also contributes to histone modification-mediated modulation of arterial BP. For example, melatonin-induced neuronal H3 acetylation elevates BP in the rostral ventrolateral medulla through an upsurge of brainstem sympathetic outflow, causing EH [106]. Renal-SNS governs high-salt-mediated hypertension. Renal-SNS activation can induce high sodium retention and low reabsorption, systemic REN secretion, and reduced renal blood flow [120, 121]. Furthermore, with-no-lysine kinase 4 (WNK4), a serine-threonine kinase, a negative regulator of Na+–Cl− cotransporter (NCC), and the epithelial Na+ channel play a key role in maintaining normal BP by decreasing sodium reabsorption in the renal distal convoluted tubule [122]. WNK4 gene downregulation and salt-sensitive hypertension in rodents [123, 124] are associated with histone modifications [125,126,127,128]. Moreover, Mu et al. reported that salt-induced histone H3 and H4 acetylation leads to glucocorticoid receptor-mediated WNK4 repression and that concurrent NCC overexpression leads to salt retention and subsequent onset of hypertension [123]. Similarly, high-salt diet-mediated hypertension in rodents is associated with lysine-specific demethylase-1 (LSD1) deficiency and associated hypermethylation of H3K9 [107].
In conclusion, similar to other epigenetic markers, histone methylation and acetylation can modulate chromatin dynamics and, hence, related gene expression. However, despite the emerging tools and techniques, the development of epigenetic histone marker-based diagnostic and prognostic methods for human EH is challenging, partly due to the extremely dynamic nature of histone modifications.
EH and miRNAs
Several noncoding RNAs (ncRNA) are implicated as epigenetic regulators of hypertension and associated target organ damage. ncRNAs (such as miRNAs, small RNAs, and long or large RNAs) act as gene regulators [129, 130]. Moreover, ncRNAs can also modulate DNA methylation and histone modifications to regulate gene expression in mammals [111, 130], thus suggesting the relationship between ncRNAs and epigenetics.
Among ncRNAs, miRNAs are evolutionarily conserved, noncoding eukaryotic RNAs consisting of ~21–26 nucleotides that can substantially influence the expression of numerous genes through mRNA degradation and translational inhibition [131, 132]. Based on rodent and human studies, numerous miRNAs have been implicated as regulators of the CV system [133, 134], as well as modulators of arterial BP [135], thus emerging as potential biomarkers and therapeutic targets in various CV diseases, including EH.
Examples of miRNA contributions to EH
As mentioned above, among the major BP-regulating pathways, RAAS is a well-known pathway in which angiotensin II regulates fluid homeostasis and BP by aldosterone stimulation. RAAS-regulated AGT gene-mediated expression of microRNA-21 (miR-21) induces aldosterone secretion in human adrenocortical cells under in vitro conditions, suggesting the possible role of miR-21 in hypertension in humans [136]. Accordingly, evidence shows the close association between miR-21 and hypertension-mediated target organ damage [137]. Both miR-27a and miR-27b downregulate ACE1 gene expression, while miR-330 is reduced to upregulate angiotensin II type-2 (AT2) receptor gene translation to alter the RAAS pathway in the malnourished fetal brain [72]. Similarly, an in vitro study using HUVECs showed that miR-155 downregulates AGTR1 mRNA, which is associated with severe EH [138]. Another human study reported that miR-181a and miR-663 targeting REN mRNA were under-expressed in the kidneys of an EH patient [139]. Robertson et al. showed that miR-24 regulates the expression of aldosterone synthase genes CYP11B1 and CYP11B2, along with the production of cortisol and aldosterone [59], which, as previously noted, have an essential role in BP regulation.
In addition, using an in vitro system, miR-124 and miR-135a have been shown to target mineralocorticoid receptor, nuclear receptor subfamily 3 group C member 2 (NR3C2), a gene associated with RAAS that is involved in familial hypertension [140]. Moreover, a genetically hypertensive mouse model (BPH/2J) and hypertensive patients show downregulation of miR-181a and concurrent overexpression of REN [139, 141]. In response to a high-salt diet, SHR model studies revealed that downregulation of renal miR-24 and miR-34a and upregulation of their target uncoupling protein 2 (UCP2) gene and protein expression may help to prevent EH and associated oxidative renal damage [142]. Ling et al. reported the correlation between hypertension and miR-320 [which targets insulin growth factor-1 receptor (IGF1R)], miR-26b [which targets phosphatase and tensin homolog (PTEN)], and miR-21 (which targets PTEN) in salt-induced arterial hypertensive rats [143]. Another human study revealed that miRNAs associated with arterial remodeling are closely related to EH [144]. In contrast to healthy subjects, a hypertensive patient’s PBMCs demonstrated downregulation of miR-133, miR-143, and miR-145, but upregulation of miR-1 and miR-21, in association with abnormal BP [144]. Moreover, European population studies have shown that NPPA, the gene expressing ANP, is correlated with BP regulation in humans. In response to a high-salt diet, miR-425 downregulates NPPA, leading to ANP underproduction and salt overload during hypertension [145]. In addition, placentas from preeclamptic pregnancies show 34 differentially expressed miRNAs, including miR-210 and miR-155 [146], which may contribute to the pre-eclampsia phenotype. In HUVECs obtained from pregnant women with severe pre-eclampsia, this is further explained by the report suggesting that AGTR1 expression regulating miR-155 is downregulated, thus influencing BP levels [138]. Recently, Wang et al. identified several miRNAs and their target genes in a hypertensive patient’s kidney, including miR-21 [targeting collagen, type XII, alpha 1 (COL12A1) and asporin (ASPN)], miR-30e, and miR-374b [targeting sodium voltage-gated channel alpha subunit 2 (SCN2A)] [147]. Recently, Zhang et al. demonstrated that miR-199a, miR-208a, and miR-122 are associated with human hypertension and that dysregulation of these miRNAs could increase the risk of hypertension [148]. Another study in South African women showed a significant decrease in miR-222 and an increase in miR-29a and 181a during pre-eclampsia [149]. Fanfan et al. reported that miR-506 is elevated in the peripheral blood of hypertensive patients and is associated with vascular endothelial injury by targeting Beclin1 (BECN1) expression [150]. Similarly, Krishnan et al. observed the upregulation of miR-510 in the peripheral blood of hypertensive patients, which may be regulated by DNA methylation [151]. In addition, Liao et al. reported the significant upregulation of BP-regulating L-type voltage-gated Ca2+ channel (LTCC) α1C subunit expression in SHR hypertensive arteries with concomitant downregulation of miR-328 [152], demonstrating the epigenetic regulation of EH.
Overall, complex genetic and metabolic signaling during EH suggests that numerous miRNAs may be involved in the regulation of key genes. Thus, based on current understanding, miRNAs may be used as novel biomarkers for EH pathogenesis, with future clinical implications.
Conclusion and future direction
As suggested by the above-listed evidence (Table 1) and different databases (Supplementary data 1), EH is a polygenic disease, and most of the EH-related genes are finely regulated via different epigenetic mechanisms, such as DNA methylation, post-translational histone modifications, and miRNAs. In light of the current literature, it is worth mentioning that most of the EH-related organs/cells and genes are affected by one or more types of epigenetic regulation. For example, most of the genes related to EH have CpG sites (DNA methylation) and are targeted by different miRNAs, and they may or may not be affected by other epigenetic markers (Fig. 4; Supplementary data 1). Overall, the combined effect of all these epigenetic changes appears to indicate tremendous potential of the growth of the field of EH diagnostics and therapeutics.
In agreement, utilizing cutting-edge technologies such as large-scale GWAS and EWAS, it is now possible to test numerous genetic and epigenetic variants across the human genome in any disorder, including EH. Using the same approach, several novel BP loci have been reported [17], and additional data are expected to add more common/rare variants to the recent literature to further enhance our current understanding. Similarly, the rapid growth of the modern epigenetic field has guided us toward several other EH-associated heritable variations. Therefore, the combined mechanistic awareness of genetic and epigenetic regulators will aid in the exploration of EH pathogenesis as scientists search for new therapeutic targets to develop innovative future personalized medicine to achieve promising clinical outcomes. Furthermore, the ability to explore genomic data through the use of new advanced techniques, such as whole genome and exome sequencing, can aid in efficiently examining the genomic regulatory regions and associated epigenetic markers affecting variants involved in EH. These approaches will also provide the significant potential to explore beyond the single variant risk model to simultaneously study multiple risk variants of EH. Overall, a clear understanding of EH risk factors in the form of genetic and epigenetic markers will be critical for the development of patient-centric and personalized approaches to therapy.
References
Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2224–60.
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M et al., American Heart Association Statistics C, Stroke Statistics S. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–322.
Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med. 2011;17:1402–9.
Parati G, Ochoa JE, Lombardi C, Bilo G. Assessment and management of bloodpressure variability. Nat Rev Cardiol. 2013;10:143–55.
Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–56.
Martinez-Aguayo A, Fardella C. Genetics of hypertensive syndrome. Horm Res. 2009;71:253–9.
Franceschini N, Le TH. Genetics of hypertension: discoveries from the bench to human populations. Am J Physiol Ren Physiol. 2014;306:F1–11.
Morgado J, Sanches B, Anjos R, Coelho C. Programming of essential hypertension: what pediatric cardiologists need to know. Pediatr Cardiol. 2015;36:1327–37.
Griffin KA. Hypertensive kidney injury and the progression of chronic kidney disease. Hypertension. 2017;70:687–94.
Gasecki D, Kwarciany M, Nyka W, Narkiewicz K. Hypertension, brain damage and cognitive decline. Curr Hypertens Rep. 2013;15:547–58.
Munroe PB, Barnes MR, Caulfield MJ. Advances in blood pressure genomics. Circ Res. 2013;112:1365–79.
Timberlake DS, O'Connor DT, Parmer RJ. Molecular genetics of essential hypertension: recent results and emerging strategies. Curr Opin Nephrol Hypertens. 2001;10:71–9.
Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41:677–87.
Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L et al. Genomewide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–76.
Kato N, Takeuchi F, Tabara Y, Kelly TN, Go MJ, Sim X et al. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet. 2011;43:531–8.
Kelly TN, Takeuchi F, Tabara Y, Edwards TL, Kim YJ, Chen P et al. Genome-wide association study meta-analysis reveals transethnic replication of mean arterial and pulse pressure loci. Hypertension. 2013;62:853–9.
Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50:1412–25.
Loscalzo J, Handy DE Epigenetic modifications: basic mechanisms and role in cardiovascular disease (2013 Grover Conference series). Pulm Circ. 2014; 4:169–74.
Wise IA, Charchar FJ. Epigenetic modifications in essential hypertension. Int J Mol Sci. 2016;17:451.
Lopez-Jaramillo P, Camacho PA, Forero-Naranjo L. The role of environment and epigenetics in hypertension. Expert Rev Cardiovasc Ther. 2013;11:1455–7.
Kato N, Loh M, Takeuchi F, Verweij N, Wang X, Zhang W et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat Genet. 2015;47:1282–93.
Stephens KE, Miaskowski CA, Levine JD, Pullinger CR, Aouizerat BE. Epigenetic regulation and measurement of epigenetic changes. Biol Res Nurs. 2013;15:373–81.
Raftopoulos L, Katsi V, Makris T, Tousoulis D, Stefanadis C, Kallikazaros I. Epigenetics, the missing link in hypertension. Life Sci. 2015;129:22–6.
Wang X, Snieder H. Genome-wide association studies and beyond: what's next in blood pressure genetics? Hypertension. 2010;56:1035–7.
Fagard RH. Exercise is good for your blood pressure: effects of endurance training and resistance training. Clin Exp Pharmacol Physiol. 2006;33:853–6.
Ash GI, Eicher JD, Pescatello LS. The promises and challenges of the use of genomics in the prescription of exercise for hypertension: the 2013 update. Curr Hypertens Rev. 2013;9:130–47.
Rakyan VK, Hildmann T, Novik KL, Lewin J, Tost J, Cox AV et al. DNA methylation profiling of the human major histocompatibility complex: a pilot study for the human epigenome project. PLoS Biol. 2004;2:e405.
Abbott A. Project set to map marks on genome. Nature. 2010;463:596–7.
Feinberg AP. Epigenetics at the epicenter of modern medicine. JAMA. 2008;299:1345–50.
Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–22.
Millis RM. Epigenetics and hypertension. Curr Hypertens Rep. 2011;13:21–8.
Ballestar E, Wolffe AP. Methyl-CpG-binding proteins. Targeting specific gene repression. Eur J Biochem. 2001;268:1–6.
Ballestar E, Paz MF, Valle L, Wei S, Fraga MF, Espada J et al. Methyl-CpG binding proteins identify novel sites of epigenetic inactivation in human cancer. EMBO J. 2003;22:6335–45.
Terry MB, Delgado-Cruzata L, Vin-Raviv N, Wu HC, Santella RM. DNA methylation in white blood cells: association with risk factors in epidemiologic studies. Epigenetics. 2011;6:828–37.
Riviere G, Lienhard D, Andrieu T, Vieau D, Frey BM, Frey FJ. Epigenetic regulation of somatic angiotensin-converting enzyme by DNA methylation and histone acetylation. Epigenetics. 2011;6:478–89.
Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–13.
Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21:5400–13.
Baccarelli A, Rienstra M, Benjamin EJ. Cardiovascular epigenetics: basic concepts and results from animal and human studies. Circ Cardiovasc Genet. 2010;3:567–73.
Udali S, Guarini P, Moruzzi S, Choi SW, Friso S. Cardiovascular epigenetics: from DNA methylation to microRNAs. Mol Asp Med. 2013;34:883–901.
Globisch D, Munzel M, Muller M, Michalakis S, Wagner M, Koch S et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 2010;5:e15367.
Jones PA. The DNA methylation paradox. Trends Genet. 1999;15:34–7.
Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–3.
Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. 2004;32:4100–8.
Branco MR, Ficz G, Reik W. Uncovering the role of 5-ydroxymethylcytosine in the epigenome. Nat Rev Genet. 2011;13:7–13.
Song CX, Yi C, He C. Mapping recently identified nucleotide variants in the genome and transcriptome. Nat Biotechnol. 2012;30:1107–16.
Liu Y, Liu P, Yang C, Cowley AW Jr., Liang M. Base-resolution maps of 5-methylcytosine and 5-hydroxymethylcytosine in Dahl S rats: effect of salt and genomic sequence. Hypertension. 2014;63:827–38.
Smolarek I, Wyszko E, Barciszewska AM, Nowak S, Gawronska I, Jablecka A et al. Global DNA methylation changes in blood of patients with essential hypertension. Med Sci Monit. 2010;16:CR149–55.
Kulkarni A, Chavan-Gautam P, Mehendale S, Yadav H, Joshi S. Global DNA methylation patterns in placenta and its association with maternal hypertension in pre-eclampsia. DNA Cell Biol. 2011;30:79–84.
Wang X, Falkner B, Zhu H, Shi H, Su S, Xu X et al. A genome-wide methylation study on essential hypertension in young African American males. PLoS ONE. 2013;8:e53938.
Assadi F. Relation of left ventricular hypertrophy to microalbuminuria and Creactive protein in children and adolescents with essential hypertension. Pediatr Cardiol. 2008;29:580–4.
Stenzig J, Schneeberger Y, Loser A, Peters BS, Schaefer A, Zhao RR et al. Pharmacological inhibition of DNA methylation attenuates pressure overload-induced cardiac hypertrophy in rats. J Mol Cell Cardiol. 2018;120:53–63.
Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol. 2007;213:384–90.
Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–30.
Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905.
Wen L, Li X, Yan L, Tan Y, Li R, Zhao Y et al. Wholegenome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. 2014;15:R49.
Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X et al. Selective chemical labeling reveals the genome-wide distribution of 5- hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72.
Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–91.
Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068–70.
Robertson S, MacKenzie SM, Alvarez-Madrazo S, Diver LA, Lin J, Stewart PM et al. MicroRNA-24 is a novel regulator of aldosterone and cortisol production in the human adrenal cortex. Hypertension. 2013;62:572–8.
Fardella CE, Mosso L, Gomez-Sanchez C, Cortes P, Soto J, Gomez L et al. Primary hyperaldosteronism in essential hypertensives: prevalence, biochemical profile, and molecular biology. J Clin Endocrinol Metab. 2000;85:1863–7.
Friso S, Pizzolo F, Choi SW, Guarini P, Castagna A, Ravagnani V et al. Epigenetic control of 11 beta-hydroxysteroid dehydrogenase 2 gene promoter is related to human hypertension. Atherosclerosis. 2008;199:323–7.
Baserga M, Kaur R, Hale MA, Bares A, Yu X, Callaway CW et al. Fetal growth restriction alters transcription factor binding and epigenetic mechanisms of renal 11beta-hydroxysteroid dehydrogenase type 2 in a sexspecific manner. Am J Physiol Regul Integr Comp Physiol. 2010;299:R334–42.
Zhao Y, Gong X, Chen L, Li L, Liang Y, Chen S et al. Site-specific methylation of placental HSD11B2 gene promoter is related to intrauterine growth restriction. Eur J Hum Genet. 2014;22:734–40.
Takeda Y, Demura M, Wang F, Karashima S, Yoneda T, Kometani M et al. Epigenetic regulation of aldosterone synthase gene by sodium and angiotensin II. J Am Heart Assoc. 2018;7:e008281.
Lee HA, Baek I, Seok YM, Yang E, Cho HM, Lee DY et al. Promoter hypomethylation upregulates Na+-K+-2Cl- cotransporter 1 in spontaneously hypertensive rats. Biochem Biophys Res Commun. 2010;396:252–7.
Cho HM, Lee HA, Kim HY, Han HS, Kim IK. Expression of Na + -K + -2Clcotransporter 1 is epigenetically regulated during postnatal development of hypertension. Am J Hypertens. 2011;24:1286–93.
Ferrario CM. Role of angiotensin II in cardiovascular disease therapeutic implications of more than a century of research. J Renin Angiotensin Aldosterone Syst. 2006;7:3–14.
Wang F, Demura M, Cheng Y, Zhu A, Karashima S, Yoneda T et al. Dynamic CCAAT/enhancer binding protein-associated changes of DNA methylation in the angiotensinogen gene. Hypertension. 2014;63:281–8.
Calhoun DA. Aldosterone and cardiovascular disease: smoke and fire. Circulation. 2006;114:2572–4.
Bogdarina I, Welham S, King PJ, Burns SP, Clark AJ. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ Res. 2007;100:520–6.
Fan R, Mao SQ, Gu TL, Zhong FD, Gong ML, Hao LM et al. Preliminary analysis of the association between methylation of the ACE2 promoter and essential hypertension. Mol Med Rep. 2017;15:3905–11.
Goyal R, Goyal D, Leitzke A, Gheorghe CP, Longo LD. Brain renin-angiotensin system: fetal epigenetic programming by maternal protein restriction during pregnancy. Reprod Sci. 2010;17:227–38.
Litwin M, Michalkiewicz J, Trojanek J, Niemirska A, Wierzbicka A, Szalecki M. Altered genes profile of renin-angiotensin system, immune system, and adipokines receptors in leukocytes of children with primary hypertension. Hypertension. 2013;61:431–6.
Fan R, Mao S, Zhong F, Gong M, Yin F, Hao L et al. Association of AGTR1 promoter methylation levels with essential hypertension risk: a matched case- control study. Cytogenet Genome Res. 2015;147:95–102.
Lin J, Lin S, Wu Y, Wang X, Wu S, Li H. Hypomethylation of the angiotensin II type I receptor (AGTR1) gene along with environmental factors increases the risk for essential hypertension. Cardiology. 2017;137:126–35.
Pei F, Wang X, Yue R, Chen C, Huang J, Huang J et al. Differential expression and DNA methylation of angiotensin type 1A receptors in vascular tissues during genetic hypertension development. Mol Cell Biochem. 2015;402:1–8.
den Ouden DT, Meinders AE. Vasopressin: physiology and clinical use in patients with vasodilatory shock: a review. Neth J Med. 2005;63:4–13.
Auger CJ, Coss D, Auger AP, Forbes-Lorman RM. Epigenetic control of vasopressin expression is maintained by steroid hormones in the adult male rat brain. Proc Natl Acad Sci USA. 2011;108:4242–7.
Greenwood MP, Greenwood M, Gillard BT, Loh SY, Paton JF, Murphy D. Epigenetic control of the vasopressin promoter explains physiological ability to regulate vasopressin transcription in dehydration and salt loading states in the rat. J Neuroendocrinol. 2016;28:4.
Cowley AW, Jr. Cushman WC, Quillen EW, Jr. Skelton MM, Langford HG. Vasopressin elevation in essential hypertension and increased responsiveness to sodium intake. Hypertension. 1981;3(3 Pt 2):I93–100.
Morton JJ, Padfield PL. Vasopressin and hypertension in man. J Cardiovasc Pharmacol. 1986;8 Suppl 7:S101–6.
Mao SQ, Fan R, Gu TL, Zhong QL, Gong ML, Dong CZ et al. Hypermethylation of SCNN1A gene-body increases the risk of essential hypertension. Int J Clin Exp Pathol. 2016;9:8047–56.
Zhong Q, Liu C, Fan R, Duan S, Xu X, Zhao J et al. Association of SCNN1B promoter methylation with essential hypertension. Mol Med Rep. 2016;14:5422–8.
Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–4.
Cirino G, Fiorucci S, Sessa WC. Endothelial nitric oxide synthase: the Cinderella of inflammation? Trends Pharmacol Sci. 2003;24:91–5.
Li Q, Youn JY, Cai H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J Hypertens. 2015;33:1128–36.
Gao L, Chalupsky K, Stefani E, Cai H. Mechanistic insights into folic aciddependent vascular protection: Dihydrofolate reductase (DHFR)-mediated reduction in oxidant stress in endothelial cells and angiotensin II-infused mice: a novel HPLC-based fluorescent assay for DHFR activity. J Mol Cell Cardiol. 2009;47:752–60.
Chan Y, Fish JE, D'Abreo C, Lin S, Robb GB, Teichert AM et al. The cell-specific expression of endothelial nitric-oxide synthase: a role for DNA methylation. J Biol Chem. 2004;279:35087–100.
Iravanian S, Dudley SC, Jr. The renin-angiotensin-aldosterone system (RAAS) and cardiac arrhythmias. Heart Rhythm. 2008;5 Suppl 6:S12–7.
Kim YM, Guzik TJ, Zhang YH, Zhang MH, Kattach H, Ratnatunga C et al. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ Res. 2005;97:629–36.
Dasgupta C, Chen M, Zhang H, Yang S, Zhang L. Chronic hypoxia duringgestation causes epigenetic repression of the estrogen receptor-alpha gene in ovine uterine arteries via heightened promoter methylation. Hypertension. 2012;60:697–704.
Meems LM, Mahmud H, Buikema H, Tost J, Michel S, Takens J et al. Parental vitamin D deficiency during pregnancy is associated with increased blood pressure in offspring via Panx1 hypermethylation. Am J Physiol Heart Circ Physiol. 2016;311:H1459–69.
Mannelli M, Pupilli C, Lanzillotti R, Ianni L, Serio M. Catecholamines and blood pressure regulation. Horm Res. 1990;34:156–60.
Esler M, Eikelis N, Schlaich M, Lambert G, Alvarenga M, Kaye D et al. Human sympathetic nerve biology: parallel influences of stress and epigenetics in essential hypertension and panic disorder. Ann N Y Acad Sci. 2008;1148:338–48.
Higuchi T, Kanzaki H, Nakayama H, Fujimoto M, Hatayama H, Kojima K et al. Induction of tissue inhibitor of metalloproteinase 3 gene expression during in vitro decidualization of human endometrial stromal cells. Endocrinology. 1995;136:4973–81.
Chelbi ST, Mondon F, Jammes H, Buffat C, Mignot TM, Tost J et al. Expressional and epigenetic alterations of placental serine protease inhibitors—SERPINA3 is a potential marker of preeclampsia. Hypertension. 2007;49:76–83.
Gascoin-Lachambre G, Buffat C, Rebourcet R, Chelbi ST, Rigourd V, Mondon F et al. Cullins in human intrauterine growth restriction: expressional and epigenetic alterations. Placenta. 2010;31:151–7.
Zhang LN, Liu PP, Wang L, Yuan F, Xu L, Xin Y et al. Lower ADD1 gene promoter DNA methylation increases the risk of essential hypertension. PLoS ONE. 2013;8:e63455.
Bayoumy NMK, El-Shabrawi MM, Leheta OF, Omar HH. alpha-Adducin gene promoter DNA methylation and the risk of essential hypertension. Clin Exp Hypertens. 2017;39:764–8.
Jin F, Li X, Wang Z, Liu Y, Liu J, Sun D et al. Association of mitofusin 2 methylation and essential hypertension: a case-control study in a Chinese population. Hypertens Res. 2018;41:605–13.
Mao SQ, Sun JH, Gu TL, Zhu FB, Yin FY, Zhang LN. Hypomethylation of interleukin-6 (IL-6) gene increases the risk of essential hypertension: a matched case-control study. J Hum Hypertens. 2017;31:530–6.
Mao S, Gu T, Zhong F, Fan R, Zhu F, Ren P et al. Hypomethylation of the Toll-like receptor-2 gene increases the risk of essential hypertension. Mol Med Rep. 2017;16:964–70.
Bao XJ, Mao SQ, Gu TL, Zheng SY, Zhao JS, Zhang LN. Hypomethylation of the interferon gamma gene as a potential risk factor for essential hypertension: a case-control study. Tohoku J Exp Med. 2018;244:283–90.
Fan R, Wang WJ, Zhong QL, Duan SW, Xu XT, Hao LM et al. Aberrant methylation of the GCK gene body is associated with the risk of essential hypertension. Mol Med Rep. 2015;12:2390–4.
Rodriguez-Iturbe B. Arteriolar remodeling in essential hypertension: are connective tissue growth factor and transforming growth factor involved? Kidney Int. 2006;69:1104–5.
Irmak MK, Sizlan A. Essential hypertension seems to result from melatonininduced epigenetic modifications in area postrema. Med Hypotheses. 2006;66:1000–7.
Pojoga LH, Williams JS, Yao TM, Kumar A, Raffetto JD, do Nascimento GRA et al. Histone demethylase LSD1 deficiency during high-salt diet is associated with enhanced vascular contraction, altered NO-cGMP relaxation pathway, and hypertension. Am J Physiol-Heart Circ Physiol. 2011;301:H1862–71.
Papait R, Cattaneo P, Kunderfranco P, Greco C, Carullo P, Guffanti A et al. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc Natl Acad Sci USA. 2013;110:20164–9.
Samavat S, Ahmadpoor P, Samadian F. Aldosterone, hypertension, and beyond. Iran J Kidney Dis. 2011;5:71–6.
Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100.
Frias-Lasserre D, Villagra CA. The Importance of ncRNAs as epigenetic mechanisms in phenotypic variation and organic evolution. Front Microbiol. 2017;8:2483.
Tabuchi TM, Rechtsteiner A, Jeffers TE, Egelhofer TA, Murphy CT, Strome S. Caenorhabditis elegans sperm carry a histone-based epigenetic memory of both spermatogenesis and oogenesis. Nat Commun. 2018;9:4310.
Lee HA, Cho HM, Lee DY, Kim KC, Han HS, Kim IK. Tissue-specific upregulation of angiotensin-converting enzyme 1 in spontaneously hypertensive rats through histone code modifications. Hypertension. 2012;59:621–6.
Wang J, Yin N, Deng Y, Wei Y, Huang Y, Pu X et al. Ascorbic acid protects against hypertension through downregulation of ACE1 gene expression mediated by histone deacetylation in prenatal inflammation-induced offspring. Sci Rep. 2016;6:39469.
Fish JE, Matouk CC, Rachlis A, Lin S, Tai SC, D'Abreo C et al. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J Biol Chem. 2005;280:24824–38.
Cho HM, Lee DY, Kim HY, Lee HA, Seok YM, Kim IK. Upregulation of the Na(+)-K(+)-2Cl(-) cotransporter 1 via histone modification in the aortas of angiotensin IIinduced hypertensive rats. Hypertens Res. 2012;35:819–24.
Duarte JD, Zineh I, Burkley B, Gong Y, Langaee TY, Turner ST et al. Effects of genetic variation in H3K79 methylation regulatory genes on clinical blood pressure and blood pressure response to hydrochlorothiazide. J Transl Med. 2012;10:56.
Zhang D, Yu ZY, Cruz P, Kong Q, Li S, Kone BC. Epigenetics and the control of epithelial sodium channel expression in collecting duct. Kidney Int. 2009;75:260–7.
Mehrotra A, Joe B, de la Serna IL. SWI/SNF chromatin remodeling enzymes are associated with cardiac hypertrophy in a genetic rat model of hypertension. J Cell Physiol. 2013;228:2337–42.
Guild SJ, Eppel GA, Malpas SC, Rajapakse NW, Stewart A, Evans RG. Regional responsiveness of renal perfusion to activation of the renal nerves. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1177–86.
DiBona GF. Physiology in perspective: the Wisdom of the Body. Neural control of the kidney. Am J Physiol Regul Integr Comp Physiol. 2005;289:R633–41.
Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR et al. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet. 2006;38:1124–32.
Mu S, Shimosawa T, Ogura S, Wang H, Uetake Y, Kawakami-Mori F et al. Epigenetic modulation of the renal betaadrenergic-WNK4 pathway in salt-sensitive hypertension. Nat Med. 2011;17:573–80.
Acres OW, Satou R, Navar LG, Kobori H. Contribution of a nuclear factor-kappaB binding site to human angiotensinogen promoter activity in renal proximal tubular cells. Hypertension. 2011;57:608–13.
Li C, Li Y, Li Y, Liu H, Sun Z, Lu J et al. Glucocorticoid repression of human with-no-lysine (K) kinase-4 gene expression is mediated by the negative response elements in the promoter. J Mol Endocrinol. 2008;40:3–12.
Abrahams JM, Lenart CJ, Tobias ME. Temporal variation of induction neurogenesis in a rat model of transient middle cerebral artery occlusion. Neurol Res. 2009;31:528–33.
Fujita T. Mechanism of salt-sensitive hypertension: focus on adrenal and sympathetic nervous systems. J Am Soc Nephrol. 2014;25:1148–55.
Akechi T, Momino K, Yamashita T, Fujita T, Hayashi H, Tsunoda N et al. Contribution of problem-solving skills to fear of recurrence in breast cancer survivors. Breast Cancer Res Treat. 2014;145:205–10.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74.
Costa FF. Non-coding RNAs, epigenetics and complexity. Gene. 2008;410:9–17.
Morris KV. siRNA-mediated transcriptional gene silencing: the potential mechanism and a possible role in the histone code. Cell Mol Life Sci. 2005;62:3057–66.
Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110.
Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469:336–42.
Arif M, Pandey R, Alam P, Jiang S, Sadayappan S, Paul A et al. MicroRNA-210-mediated proliferation, survival, and angiogenesis promote cardiac repair post myocardial infarction in rodents. J Mol Med (Berl). 2017;95:1369–85.
Nguyen Dinh Cat A, Ouvrard-Pascaud A, Tronche F, Clemessy M, Gonzalez- Nunez D, Farman N et al. Conditional transgenic mice for studying the role of the glucocorticoid receptor in the renal collecting duct. Endocrinology. 2009;150:2202–10.
Romero DG, Plonczynski MW, Carvajal CA, Gomez-Sanchez EP, Gomez-Sanchez CE. Microribonucleic acid-21 increases aldosterone secretion and proliferation in H295R human adrenocortical cells. Endocrinology. 2008;149:2477–83.
Li X, Wei Y, Wang Z. microRNA-21 and hypertension. Hypertens Res. 2018;41:649–61.
Cheng W, Liu T, Jiang F, Liu C, Zhao X, Gao Y et al. microRNA-155 regulates angiotensin II type 1 receptor expression in umbilical vein endothelial cells from severely pre-eclamptic pregnant women. Int J Mol Med. 2011;27:393–9.
Marques FZ, Campain AE, Tomaszewski M, Zukowska-Szczechowska E, Yang YH, Charchar FJ et al. Gene expression profiling reveals renin mRNA overexpression in human hypertensive kidneys and a role for microRNAs. Hypertension. 2011;58:1093–8.
Sober S, Laan M, Annilo T. MicroRNAs miR-124 and miR-135a are potential regulators of the mineralocorticoid receptor gene (NR3C2) expression. Biochem Biophys Res Commun. 2010;391:727–32.
Jackson KL, Marques FZ, Watson AMD, Palma-Rigo K, Nguyen-Huu TP, Morris BJ et al. A novel interaction between. sympathetic overactivity and aberrant regulation of renin by miR-181a in BPH/2J genetically hypertensive mice. Hypertension. 2013;62:775–81.
Di Castro S, Scarpino S, Marchitti S, Bianchi F, Stanzione R, Cotugno M et al. Differential modulation of uncoupling protein 2 in kidneys of stroke-prone spontaneously hypertensive rats under high-salt/low-potassium diet. Hypertension. 2013;61:534–41.
Ling S, Nanhwan M, Qian J, Kodakandla M, Castillo AC, Thomas B et al. Modulation of microRNAs in hypertension-induced arterial remodeling through the beta1 and beta3-adrenoreceptor pathways. J Mol Cell Cardiol. 2013;65:127–36.
Kontaraki JE, Marketou ME, Zacharis EA, Parthenakis FI, Vardas PE. Differential expression of vascular smooth muscle-modulating microRNAs in human peripheral blood mononuclear cells: novel targets in essential hypertension. J Hum Hypertens. 2014;28:510–6.
Arora P, Wu C, Khan AM, Bloch DB, Davis-Dusenbery BN, Ghorbani A et al. Atrial natriuretic peptide is negatively regulated by microRNA-425. J Clin Investig. 2013;123:3378–82.
Zhu XM, Han T, Sargent IL, Yin GW, Yao YQ. Differential expression profile of microRNAs in human placentas from preeclamptic pregnancies vs normal pregnancies. Am J Obstet Gynecol. 2009;200:661 e661–7.
Wang G, Wu L, Chen Z, Sun J. Identification of crucial miRNAs and the targets in renal cortex of hypertensive patients by expression profiles. Ren Fail. 2017;39:92–9.
Zhang X, Wang X, Wu J, Peng J, Deng X, Shen Y et al. The diagnostic values of circulating microRNAs for hypertension and bioinformatic analysis. Biosci Rep. 2018;38. pii: BSR20180525 https://www.ncbi.nlm.nih.gov/pubmed/29961674.
Khaliq OP, Murugesan S, Moodley J, Mackraj I. Differential expression of miRNAs are associated with the insulin signaling pathway in preeclampsia and gestational hypertension. Clin Exp Hypertens. 2018;40:744–751. https://www.ncbi.nlm.nih.gov/pubmed/29381395.
Yi F, Hao Y, Chong X, Zhong W. Overexpression of microRNA-506-3p aggravates the injury of vascular endothelial cells in patients with hypertension by downregulating Beclin1 expression. Exp Ther Med. 2018;15:2844–50.
Krishnan R, Mani P, Sivakumar P, Gopinath V, Sekar D. Expression and methylation of circulating microRNA-510 in essential hypertension. Hypertens Res. 2017;40:361–3.
Liao J, Zhang Y, Ye F, Zhang L, Chen Y, Zeng F et al. Epigenetic regulation of L-type voltage-gated Ca(2+) channels in mesenteric arteries of aging hypertensive rats. Hypertens Res. 2017;40:441–9.
Acknowledgements
EMU, LJM, and RCB were supported by AHA SFRN23680000 and NIH UL1 TR001425 (CTSA). MA was a postdoctoral fellowship recipient of the AHA SFRN23680000. SS was supported by NIH National Heart, Lung, and Blood grants, including R01HL130356 and R01HL105826, and the American Heart Association (AHA) Midwest Affiliate Research Programs, including Cardiovascular Genome-Phenome Study 15CVGPSD27020012 and Catalyst 17CCRG33671128.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SS provides consulting services to AstraZeneca and Amgen unrelated to the content of this manuscript. The remaining authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Arif, M., Sadayappan, S., Becker, R.C. et al. Epigenetic modification: a regulatory mechanism in essential hypertension. Hypertens Res 42, 1099–1113 (2019). https://doi.org/10.1038/s41440-019-0248-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-019-0248-0
Keywords
This article is cited by
-
RGS proteins and their roles in cancer: friend or foe?
Cancer Cell International (2023)
-
Based on network pharmacology, gastrodin attenuates hypertension-induced vascular smooth muscle cell proliferation and PI3K/AKT pathway activation
Scientific Reports (2023)
-
Maternal exercise upregulates the DNA methylation of Agtr1a to enhance vascular function in offspring of hypertensive rats
Hypertension Research (2023)
-
Novel methylation mark and essential hypertension
Journal of Genetic Engineering and Biotechnology (2022)
-
Research progress of Nedd4L in cardiovascular diseases
Cell Death Discovery (2022)
